
January is Birth Defects Awareness Month. Join the effort to raise awareness of birth defects.

There are steps you can take to be healthy before and during pregnancy.

NBDPS and BD-STEPS are large studies of birth defect risk factors supported by CDC.
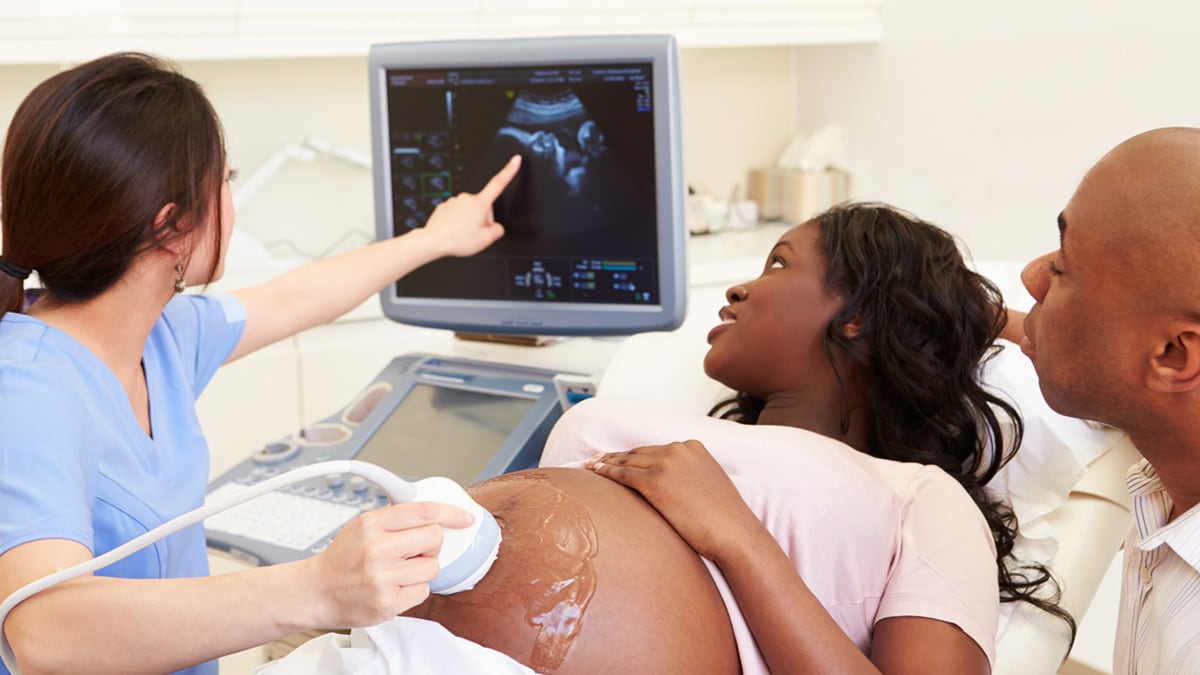
Screening tests can help identify if there may be a birth defect or other concern during pregnancy.
Learn more about birth defects

Birth defects are structural changes that are present at birth.
Specific Conditions
- Anencephaly is a birth defect in which a baby is born without parts of the brain and skull.
- Anophthalmia and microphthalmia are birth defects of a baby's eye(s).
- Anotia and microtia are birth defects of a baby's ear.
- Cleft lip and palate are birth defects that occur when a baby's lip or mouth don't form properly.
- Craniosynostosis is a birth defect in which the bones in a baby's skull join together too early.
- Diaphragmatic hernia is a birth defect where there is a hole in the diaphragm.
- Down syndrome is a condition in which a person has an extra chromosome 21.
- Encephalocele is a sac-like protrusion of the brain through an opening in the skull.
- Esophageal atresia is a birth defect of the tube (esophagus) that connects the mouth to the stomach.
- Gastroschisis is a birth defect where there is a hole in the abdominal wall beside the belly button.
- Hypospadias is a birth defect where the opening of the urethra isn't at the tip of the penis.
- Limb reduction defects occur when a part of or the entire arm or leg fails to form completely.
- Microcephaly is a birth defect where a baby's head is smaller than expected.
- Neural tube defects are severe birth defects of the brain and spine.