At a glance
All influenza viruses undergo genetic changes over time. CDC conducts year-round surveillance of circulating influenza viruses to monitor for changes in these viruses. Genome sequencing is a process that determines the order, or sequence, of the nucleotides (i.e., A, C, G and T/U) in each of the genes present in the virus's genome. An influenza virus's genome consists of all genes that make up the virus. CDC and other public health laboratories around the world sequence the gene segments of influenza viruses and contribute these genetic sequences to public databases. This allows the CDC and other researchers to compare the genes of currently circulating influenza viruses with the genes of older influenza viruses and those used in vaccines. The process of comparing genetic sequences is called genetic characterization.
Genome Sequencing
Influenza viruses are constantly changing. In fact, all influenza viruses undergo genetic changes over time. An influenza virus's genome consists of all genes that make up the virus. CDC conducts year-round surveillance of circulating influenza viruses to monitor changes in the genome of these viruses. This work is performed as part of routine U.S. influenza surveillance and as part of CDC's role as a World Health Organization (WHO) Collaborating Center for the Surveillance, Epidemiology and Control of Influenza. The information CDC collects from studying genetic changes (also known as "substitutions" or "mutations") in influenza viruses plays an important public health role by helping to determine whether vaccines and antiviral drugs will work against currently circulating influenza viruses, as well as helping to determine the potential for influenza viruses in animals to infect humans.
Genome sequencing is a process that determines the order, or sequence, of the nucleotides (i.e., A, C, G and T/U) in each of the genes present in the virus's genome. Nucleotides are organic molecules that are building blocks of nucleic acids, such as RNA and DNA. All influenza viruses consist of single-stranded RNA as opposed to dual-stranded DNA. The RNA genes of influenza viruses are made up of chains of nucleotides that are bonded together and coded by the letters A, C, G and U, which stand for adenine, cytosine, guanine, and uracil, respectively. Full genome sequencing can reveal the approximately 13,500-letter sequence of all the genes of the influenza virus's genome.
The two influenza types (A and B) that cause seasonal epidemics have eight RNA gene segments. These genes contain instructions for making new viruses and play an important role in how influenza viruses cause infection. An influenza virus's surface proteins, hemagglutinin (HA) and neuraminidase (NA), determine important properties of the virus and are included in most seasonal vaccines, which is why they are analyzed more closely. In a typical year, CDC performs whole genome sequencing on about 7,000 influenza viruses from original clinical samples collected through virologic surveillance.
Comparing the nucleotides in one gene of a virus with those of a different virus can reveal differences between the two viruses. Genetic variations are important because they can change amino acids that make up the influenza virus's proteins, resulting in structural changes to the proteins that can alter the properties of the virus. Some of these properties include the ability to evade human immunity, the ability to spread between people, and susceptibility to influenza antiviral drugs. These changes to the proteins can come in the form of amino acid substitutions, insertions, or deletions.
Genetic Characterization
CDC and other public health laboratories around the world have been sequencing the gene segments of influenza viruses since the 1980s. CDC contributes gene sequences to public databases, such as GenBank and the Global Initiative on Sharing Avian Influenza Data (GISAID), for use by researchers and public health scientists. The sequences deposited into these databases allow CDC and other researchers to compare the genes of currently circulating influenza viruses with the genes of older influenza viruses and those used in vaccines. This process of comparing genetic sequences is called genetic characterization. CDC uses genetic characterization for several reasons, including:
- To determine how closely "related," or similar, influenza viruses are to one another genetically
- To monitor how influenza viruses are evolving or changing over time
- To identify genetic changes that affect the virus's properties. For example, to identify the specific changes associated with influenza viruses that spread more easily, cause more severe disease, or develop resistance to antiviral drugs
- To assess how well a flu vaccine might protect against a particular influenza virus based on its genetic similarity to the virus
- To monitor for genetic changes in influenza viruses circulating in animal populations that could enable them to infect humans.

The genetic differences among a group of influenza viruses can be shown by organizing and displaying them in a graphic called a "phylogenetic tree." Phylogenetic trees for influenza viruses are like family (genealogy) trees for people. These trees show how closely related individual viruses are to one another. Each sequence from a specific influenza virus has its own branch on the tree. Viruses are grouped by comparing changes in nucleotides within the gene. These "nodes" where branches meet represent the common ancestor of the viruses and indicate that the viruses share similar genetic sequences. Viruses that share a common ancestor can also be described as belonging to the same clade. The degree of genetic difference (number of nucleotide differences) between viruses is represented by the length of the horizontal lines (branches) in the phylogenetic tree. The further apart viruses are on the horizontal axis of a phylogenetic tree, the more genetically different the viruses are from one another.
Phylogenetic trees of influenza viruses will usually display how similar sequences of the nucleotides for hemagglutinin (HA) genes of the vaccine virus and circulating viruses are to each other.
Genome sequencing
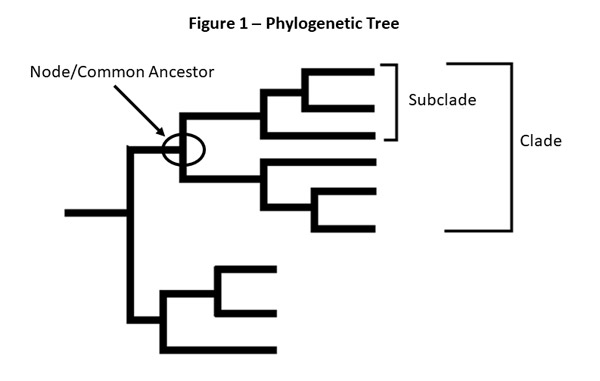
For example, after CDC sequences an influenza A(H3N2) virus collected through surveillance, the virus sequence is cataloged with other sequences that have a similar HA gene (H3) and a similar NA gene (N2). CDC compares the new virus sequence with the other virus sequences and looks for differences between them. CDC then uses a phylogenetic tree to visually represent how genetically similar the A(H3N2) viruses are to each other. In Figure 1, virus b is more genetically similar to virus c than to virus d. Viruses b and c share a common ancestor, and the total length of the horizontal branches is short.
CDC performs genetic characterization of influenza viruses year-round. These genetic data are used in conjunction with virus antigenic characterization data and other data, like human serology data, to help select the vaccine viruses for each year's flu vaccine. The analysis and selection are made twice each year to recommend vaccine viruses for both the Northern Hemisphere and the Southern Hemisphere. In the months leading up to the WHO-facilitated vaccine consultation meetings, where the recommendations are made, CDC collects influenza viruses through surveillance and compares the HA and NA gene sequences of current vaccine viruses against those of circulating flu viruses. This is one of the ways CDC assesses how closely related the circulating influenza viruses are to the viruses the seasonal flu vaccine is formulated to protect against.
Sometimes over the course of a season, circulating influenza viruses will change genetically in such a way that further analyses are needed to determine whether they remain antigenically similar to current vaccine viruses or whether a new virus needs to be included in the next flu season's vaccine. Many other data, including antigenic characterization findings and human serology data, shape the vaccine selection decisions. Antigenic characterization refers to the analysis of a virus's reaction with antibodies to help assess how it relates to the vaccine virus and other circulating influenza viruses.
Methods of Flu Genome Sequencing
Due to the constantly changing nature of influenza viruses, samples collected from a patient contain many influenza virus particles that have small genetic differences from each other. Traditionally, scientists have used a sequencing technique called "the Sanger method" to monitor influenza evolution as part of genetic characterization. Sanger sequencing identifies the predominant genetic sequence among the many influenza viruses found in a virus sample. This means small variations in the population of viruses present in a sample are not reflected in the final result. Newer technologies, such as Next Generation Sequencing (described below), are better suited for detecting small variations in the virus genes and offer advantages for whole genome sequencing.
Since 2014, CDC has been using Next Generation Sequencing (NGS) methodologies, which have greatly expanded the amount of information and detail that sequencing analysis can provide.
In a typical year, CDC performs whole genome sequencing on about 7,000 influenza viruses from original clinical specimens collected through virologic surveillance. NGS uses advanced molecular detection (AMD) to identify gene sequences from each virus in a specimen. By revealing the genetic variations among many different influenza virus particles in a single specimen, NGS can increase the speed and accuracy of sequencing each of the protein coding regions of the virus. This level of detail can directly benefit public health decision-making in important ways, but data must be carefully interpreted by highly trained experts in the context of other available information.