
Get information on the use of respirators in the workplace.

Learn more about PPE worn to minimize exposure to workplace hazards across various industries.
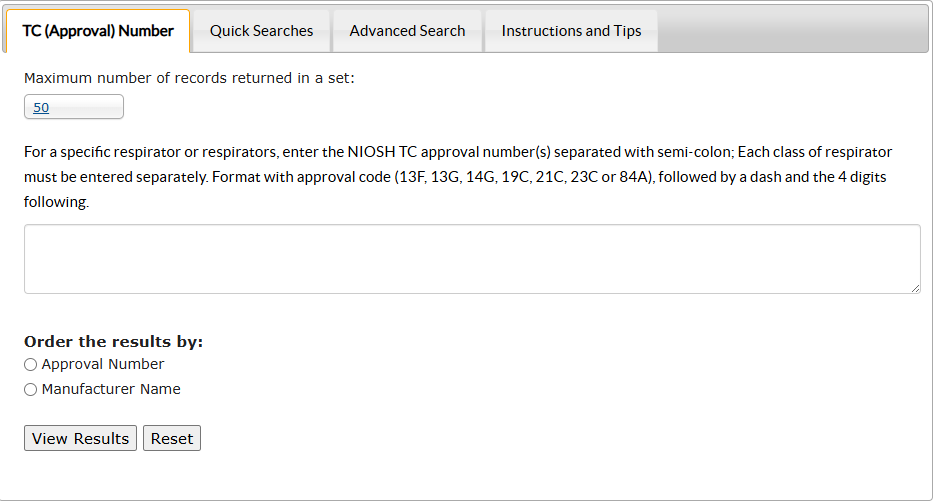

Learn about respirator fit testing
New and Noteworthy

Hazardous substances can transfer from firefighters' gear to their skin, equipment, and station.

Effective cleaning methods can protect firefighters from harmful contaminants on their gear.
NIOSH contributes to private sector standards development.
Summarizes NIOSH's findings and evaluation of three SCBAs involved in a near miss.
Shows doffing sequence to remove personal protective equipment at a fire incident.
Details the results of performance testing on fraudulent respirator filters and cartridges
Learn More

The NIOSH Respirator Approval Program is responsible for testing and approving respirators.

Find NIOSH Approved® FFRs on the CEL

Collection of NIOSH Conformity Assessment Notices and Letters.

PPE is the fifth and final level within the hierarchy of controls to reduce or remove hazards.

Information on NIOSH's respirator post-market conformity evaluations.
This webpage provides respiratory protection information and resources for the healthcare sector.