Key points
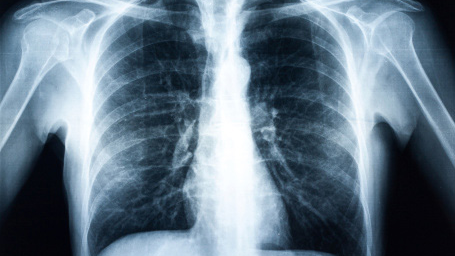
- Chest radiographs, also known as chest x-rays, are radiographic images taken of the chest.
- Digital chest radiographs help diagnose conditions affecting the lungs and are used for medical screening and surveillance.
- A B Reader is a physician trained and certified to classify chest x-ray findings of pneumoconiosis.
Overview
Chest radiography helps Identify workers with dust-induced lung diseases called pneumoconioses. Timely interventions to reduce or eliminate further dust exposure can lower disease progression.

B Readers classify radiographic images of workers exposed to mineral dust according to the International Labour Office (ILO) International Classification of Radiographs of Pneumoconioses. The ILO classification is the most widely used to evaluate pneumoconiosis in chest images. Government programs and public health agencies world-wide use classifications to characterize pneumoconiosis.
Guidelines for using chest radiographs
Using any technology requires ongoing attention to personnel, equipment, and procedures to maximize performance. Application of Digital Radiography for the Detection and Classification of Pneumoconiosis provides information on obtaining images.