Key points
- Refugee populations have unique health profiles and may be more likely to have certain health conditions less common in the general U.S. patient population.
- CDC recommends that all recently resettled refugees, regardless of age or ethnicity, have a complete blood count (CBC).
- Infants are recommended to have a newborn screening panel according to state guidelines.
- Additional, more focused laboratory testing may be warranted based on the results of these tests.
Background
The purpose of this document is to describe general testing components that do not fall into the disease-specific sections of the Domestic Medical Screening Guidance for Newly Arrived Refugees. This guidance is based on accepted best practices in refugee clinical care, with references to peer-reviewed literature.
Recommendations for Domestic Screening: Nonspecific Laboratory Tests
Refugee populations have unique health profiles, come from diverse backgrounds, and may be predisposed to certain health conditions less common in the general US patient population. Some disorders may be detected by using general, nonspecific tests. Laboratory testing may also identify those individuals at increased risk for acute and chronic health conditions.
All recently resettled refugees, regardless of age and ethnicity, should have a complete blood count (CBC) with red blood cell indices, white blood cell differential, and platelet count. These general, nonspecific tests may indicate common underlying conditions, including iron deficiency and inherited anemias. Additionally, these tests may reveal rarer conditions that if not treated or managed, may result in serious morbidity or mortality. Based on the results of these tests, additional, more focused laboratory testing may be warranted.
Commonly Encountered Hematologic Disorders in Refugees
Anemia
Anemia is common among refugees, regardless of age, sex, and ethnicity. Among African refugees resettled in Australia, a study showed that 19% of adults (n=235) had anemia, while another study showed that 37% of adult Southeast Asian refugees resettled to the United (n=521) had anemia. 12Additionally, numerous studies have reported anemia among refugee children resettled to the United States. Analyses have shown anemia in 10.7-28% of resettled refugee children, depending on age and country of origin.345
Common causes of anemia in refugee populations include primary iron deficiency, blood loss (e.g., menorrhagia, intestinal parasitosis, peri- and post-partum hemorrhage), inherited hematologic abnormalities (e.g., sickle cell disease, thalassemias, enzyme defects), and infectious diseases that cause pathology, such as bone marrow suppression, sequestration or hemolysis (e.g., malaria, visceral leishmaniasis). In many cases, the cause of anemia is multifactorial, and clinicians should consider multiple conditions if anemia is detected.6 The complete evaluation of anemia in refugees is beyond the scope of this document; common hematologic conditions encountered in refugees are discussed below.
Iron-Deficient Anemia
Iron-deficient anemia (IDA) is one of the most common causes of anemia in refugees. IDA typically manifests with microcytosis and most often affects women and children. However, all refugees are at risk.7In a convenience sample of western, central, and eastern African refugees (n=210, median age 15 years) newly arrived in Australia, 20% had ferritin levels that indicated iron deficiency.1
IDA is frequently multifactorial but is primarily caused by deficient dietary iron. See Domestic Screening Guidance for Evaluation of Nutritional Status and Growth in Refugee Children for additional information on iron-deficient anemia. Chronic blood loss, which frequently compounds iron deficiency, is commonly caused by intestinal parasitic infections, particularly hookworm. Helicobacter pylori infections may lead to gastrointestinal blood loss through ulcer formation.
Iron deficiency likely increases intestinal absorption of lead8. Given the high prevalence of iron deficiency in refugee children and pregnant and breastfeeding women, as well as the potential for lead exposure/development of elevated lead levels after US arrival, clinical management of iron deficiency should include testing for lead (see Screening for Lead during the Domestic Medical Examination for Newly Arrived Refugees for complete recommendations).
Inherited Anemias
Inherited hematologic disorders should be considered in any refugee who has anemia detected on screening, even if other potential causes exist (e.g., iron deficiency), particularly if not corrected with therapy. These disorders include a number of conditions, such as thalassemias and sickle cell disease, as well as enzyme or cell membrane defects. Most of these conditions are autosomal recessive. Therefore, it is important to both identify symptomatic refugees who are homozygous for an abnormal gene and to detect heterozygous carriers since their offspring may be affected by the disease. Of note, glucose-6-phosphate dehydrogenase (G6PD) deficiency is an X-linked recessive condition that generally does not result in chronic anemia but can result in acute hemolytic anemia after certain exposures (see below). A 2016 Nepali survey of inherited disorders found that G6PD deficiency was the most common inherited anemia, with 18% of infants and children 6-59 months of age being affected. By comparison, in the same cohort, only 2% were carriers for alpha-thalassemia, 5% had beta-thalassemia, <1% had sickle cell trait, and 0.9% had hemoglobin E.9
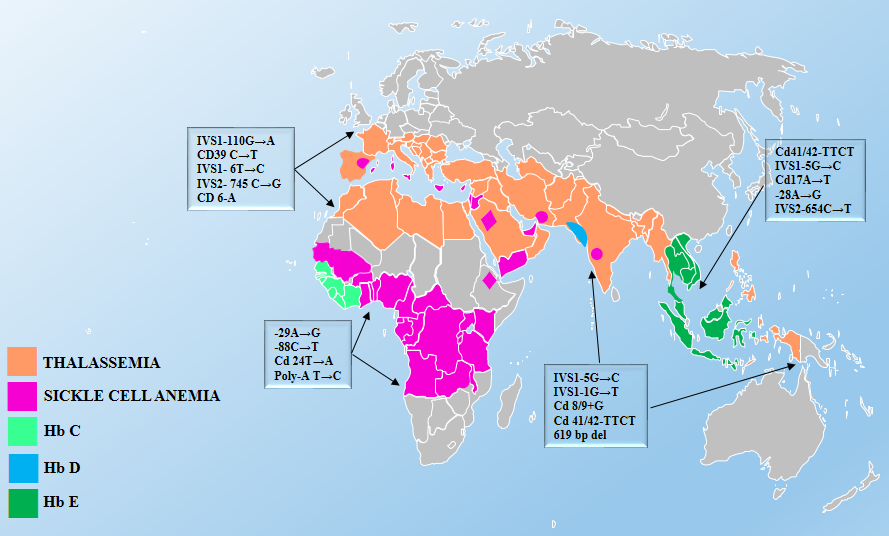
Source: Inherited Hemoglobin Disorders, edited by Anjana Munshi, ISBN 978-953-51-2198-5, Published: November 11, 2015.
Thalassemias
Thalassemias are a group of disorders characterized by a decrease in either the alpha (ɑ) or beta (β) globin chain production in red blood cells (RBCs). Globally, thalassemia is most common among people born in or descended from populations in eastern Asia, the Philippines, Indonesia, India, Pakistan, and the Middle East.10As a result of immigration from these regions, the prevalence of thalassemia has increased in North America in recent decades.10
Four conditions make up the α-thalassemias; each is defined by the number of inherited deletions of the four α-globin genes. Similarly, β-thalassemia is defined by mutations in one or both of the two β-globin genes.
Sickle Cell Disease
Sickle cell disease (SCD) is perhaps the most widely known hemoglobinopathy. Globally, 80% of people affected by sickle cell disease live in or trace their ancestry to central Africa. However, the condition may also affect people from Central and South America, the Arabian Peninsula, the Middle East, India, and the eastern Mediterranean.11 SCD is a group of inherited RBC disorders. There are many forms of the disease, with HbSS (also known as sickle cell anemia) being the most severe form. HbSC and HbS beta-thalassemia are also common types of SCD.
Anemia and other RBC abnormalities may alert the clinician to the possibility of SCD. It should be noted that individuals with sickle cell trait (SCT)—carriers of SCD due to a sickle cell mutation in only one of the two beta-globin genes—is often asymptomatic and does not cause anemia or RBC abnormalities on a routine CBC. However, individuals with SCT can pass the mutation on to their children and, under rare circumstances, SCT can cause complications. US-born infants are tested for SCD/SCT as part of their newborn screen. Clinicians should consider screening all individuals if they have anemia or red cell abnormalities that do not correct with appropriate treatment (e.g., iron supplementation). Furthermore, they should consider screening those who may be at risk of having SCT (Figure 1).
Hemoglobin E Trait
Hemoglobin E trait is frequently present in certain refugee groups, particularly among those from Southeast Asia. Both heterozygous and homozygous persons are generally asymptomatic, but may have hypochromic microcytosis and mild anemia.12However, individuals with both a hemoglobin E mutation on one of the beta-globin genes and a beta-globin gene deletion may have moderate to severe anemia. The prevalence of hemoglobin E trait is very high in parts of Southeast Asia – nearly 60% in regions of Thailand, Laos, and Cambodia.10Hemoglobin E trait is also seen in people from Indonesia, Bangladesh, northeast India, Sri Lanka, and parts of the Middle East.1213As with thalassemias, hemoglobin E red cell indices are similar to those seen in IDA. However, unless the patient is also iron deficient, administration of iron will not improve the microcytic anemia and can cause iron overload. Iron supplementation should be used cautiously in people with microcytic anemia who may have hemoglobin E trait.
Glucose-6-Phosphate Dehydrogenase Deficiency
G6PD deficiency is the most common inherited enzyme deficiency, affecting over 400 million people. In certain circumstances, G6PD may cause acute hemolytic anemia. As G6PD does not cause chronic anemia, chronic anemia would not be detected in a screening CBC unless the blood draw happens to coincide with an episode of acute hemolysis. The geographic distribution of this condition is similar to that of thalassemia, but G6PD deficiency is particularly common in Southeast Asia.11Because the gene that codes for G6PD is located on the X chromosome, males are typically more severely affected than females.
Most people with G6PD deficiency are asymptomatic until exposed to oxidizing medications or other agents (e.g., nitrofurantoin, fluoroquinolones, primaquine and tafenoquine [anti-malarials], or dapsone [commonly used for Hansen’s disease and as a prophylactic agent in HIV-infected people]); foods (e.g., fava beans); chemical exposure to naphthalene (e.g., moth balls); henna compounds; or infections that can trigger acute hemolytic anemia, which can be severe and even life-threatening. To prevent an acute hemolytic episode, any refugee from a high-risk area, particularly the Mediterranean and Southeast Asia, should be tested for G6PD activity before prescribing a medication that has been associated with an increased risk of hemolysis.14
In addition, the domestic screening should include questions to identify past episodes of hemolysis, including prolonged or unusually severe neonatal jaundice, recurrent episodes of anemia, hemoglobinuria, jaundice, or gallstones. Laboratory findings during an acute hemolytic event include normocytic anemia, increased reticulocyte count, normal liver enzymes, increased lactic dehydrogenase, decreased haptoglobin, and elevated indirect bilirubin. A urinalysis may be heme-positive without RBCs on microscopic examination.13
Eosinophilia
Eosinophilia may be defined as an eosinophil percentage exceeding 5% or an absolute eosinophil count (AEC) exceeding 400 eosinophils/mm3 (some authors use AEC of >500 eosinophils/mm3). AEC is preferred rather than using the percent of eosinophils (which some laboratories will report), as a patient will often have a normal eosinophil percentage when the AEC is elevated. When only the percentage is reported, the AEC is easily calculated by multiplying the total white blood cell count by the eosinophil percentage.
Eosinophilia in a newly arrived refugee most likely indicates a recently treated or current parasitic infection (see Presumptive Treatment and Medical Screening for Parasites in Newly Arriving Refugees), although other etiologies, such as allergies, medication reactions, and atopy, may account for the finding.
Thrombocytopenia
A variety of conditions may cause thrombocytopenia, including many infectious diseases. Hypersplenism is commonly associated with thrombocytopenia and may be caused by chronic infection with malaria or schistosomiasis (e.g., particularly in sub-Saharan African refugees from Congo).15However, many other infections can be primarily responsible (e.g., visceral leishmaniasis, brucellosis) or secondarily responsible (e.g., viral hepatitis) for the underlying splenomegaly. There are also infections that may cause thrombocytopenia through mechanisms other than splenomegaly, including HIV infection (up to 40% of infected people will have low platelet counts), Epstein-Barr virus, and viral hepatitis. The differential diagnosis of thrombocytopenia is extensive, and consultation with an expert may be needed to identify and treat the underlying cause.
Other Conditions
A CBC with differential may reveal clues to a wide range of other, less common disorders, such as malignancy (e.g., leukemia), vitamin deficiencies indicated by megaloblastic anemia (e.g., B12, folate deficiencies), anemia of chronic disease, or thyroid disease.
Newborn, Sickle Cell, and Thyroid Screening in Infants and Children
Routine newborn screening is not performed in the majority of countries from which refugees emigrate.16 If an infant is seen for a refugee medical screening, a newborn screening panel should be conducted according to state guidelines. If there are clinical concerns for an underlying medical condition in toddlers and older children, clinicians should test for the specific medical conditions in consultation with a pediatric sub-specialist.
Some experts suggest screening for hemoglobinopathies for individuals from high prevalence areas.
Thyroid disease should be considered for all infants and children (<6 years of age) as iodine deficiency and congenital hypothyroidism may occur in infants and younger children with no or minimal symptoms. Measuring thyroid-stimulating hormone (TSH) and free T4 should be considered, especially if infants have signs or symptoms of thyroid disease.
- Tiong, A.C., et al., Health issues in newly arrived African refugees attending general practice clinics in Melbourne. Med J Aust, 2006. 185(11-12): p. 602-6.
- Catanzaro, A. and R.J. Moser, Health status of refugees from Vietnam, Laos, and Cambodia. Jama, 1982. 247(9): p. 1303-8.
- Geltman, P.L., et al., Growth status and related medical conditions among refugee children in Massachusetts, 1995-1998. Am J Public Health, 2001. 91(11): p. 1800-5.
- Hayes, E.B., et al., Health status of pediatric refugees in Portland, ME. Arch Pediatr Adolesc Med, 1998. 152(6): p. 564-8.
- Yun, K., et al., Health Profiles of Newly Arrived Refugee Children in the United States, 2006-2012. Am J Public Health, 2016. 106(1): p. 128-35.
- Stauffer, W.M., D. Kamat, and P.F. Walker, Screening of international immigrants, refugees, and adoptees. Prim Care, 2002. 29(4): p. 879-905.
- Pottie, K., P. Topp, and F. Kilbertus, Case report: profound anemia. Chronic disease detection and global health disparities. Can Fam Physician, 2006. 52(3): p. 335-6.
- Wright, R.O., et al., Association between iron deficiency and blood lead level in a longitudinal analysis of children followed in an urban primary care clinic. J Pediatr, 2003. 142(1): p. 9-14.
- Ministry of Health and Population, Nepal National Micronutrient Status Survey. 2016, Government of Nepal, USAID, CDC, EU, New ERA, unicef
- Vichinsky, E.P., Changing patterns of thalassemia worldwide. Ann N Y Acad Sci, 2005. 1054: p. 18-24.
- Theodorsson, E., H. Birgens, and T.A. Hagve, Haemoglobinopathies and glucose-6-phosphate dehydrogenase deficiency in a Scandinavian perspective. Scand J Clin Lab Invest, 2007. 67(1): p. 3-10.
- Rees, D.C., et al., The hemoglobin E syndromes. Ann N Y Acad Sci, 1998. 850: p. 334-43.
- Jeng, M.R. and E. Vichinsky, Hematologic problems in immigrants from Southeast Asia. Hematol Oncol Clin North Am, 2004. 18(6): p. 1405-22, x.
- Glader, B., Diagnosis and management of glucose-6-phosphate dehydrogenase (G6PD) deficiency, Partial list of medicines and other substances thought to be unsafe or safe in individuals with G6PD deficiency, Editor. 2022, UpToDate.
- Zambrano, L.D., et al., Unresolved Splenomegaly in Recently Resettled Congolese Refugees – Multiple States, 2015-2018. MMWR Morb Mortal Wkly Rep, 2018. 67(49): p. 1358-1362.
- Therrell, B.L., et al., Current status of newborn screening worldwide: 2015. Semin Perinatol, 2015. 39(3): p. 171-87.