Virology
Hantaviruses
Hantaviruses belong to the bunyavirus family of viruses. There are 5 genera within the family: bunyavirus, phlebovirus, nairovirus, tospovirus, and hantavirus. Each is made up of negative-sensed, single-stranded RNA viruses. All these genera include arthropod-borne viruses, with the exception of hantavirus, which is rodent-borne.
Like other members of the bunyavirus family, hantaviruses are enveloped viruses with a genome that consists of three single-stranded RNA segments designated S (small), M (medium), and L (large). All hantaviral genes are encoded in the negative (genome complementary) sense. The S RNA encodes the nucleocapsid (N) protein. The M RNA encodes a polyprotein that is cotranslationally cleaved to yield the envelope glycoproteins G1 and G2. The L RNA encodes the L protein, which functions as the viral transcriptase/replicase. Within virions, the genomic RNAs of hantaviruses are thought to complex with the N protein to form helical nucleocapsids, which circularize due to sequence complementarity between the 5′ and 3′ terminal sequences of each genomic segment.
Hantaviruses replicate exclusively in the host cell cytoplasm. Entry into host cells is thought to occur by attachment of virions to cellular receptors and subsequent endocytosis. Nucleocapsids are introduced into the cytoplasm by pH-dependent fusion of the virion with the endosomal membrane. Transcription of viral genes is initiated by association of the L protein with the three nucleocapsid species. In addition to transcriptase and replicase functions, the viral L protein is also thought to have an endonuclease activity that cleaves cellular messenger RNAs (mRNAs) for the production of capped primers used to initiate transcription of viral mRNAs. As a result of this “cap snatching”, the mRNAs of hantaviruses are believed to be capped and contain nontemplated 5′ terminal extensions. The viral N and L mRNAs are thought to undergo translation at free ribosomes, whereas the M mRNA is translated in the endoplasmic reticulum. G1 and G2 glycoproteins form heterodimers and are then transported from the endoplasmic reticulum to the Golgi complex, where glycosylation is completed. The L protein produces nascent genomes by replication via a positive-sense RNA intermediate. Hantavirions are believed to form by association of nucleocapsids with glycoproteins embedded in the membranes of the Golgi, followed by budding into the Golgi cisternae. Nascent virions are then transported in secretory vesicles to the plasma membrane and released by exocytosis.
Hantaviruses Causing HPS
Sin Nombre virus (SNV) was first isolated from rodents collected on the premises of one of the initial HPS patients in the Four Corners region. Isolation was achieved through blind passage in Peromyscus maniculatus and subsequent adaptation to growth in Vero E6 cells. Additional viral strains have also been isolated from P. maniculatus associated with a fatal case in California and P. leucopus from the vicinity of probable infection of a New York case. Black Creek Canal virus was isolated from S. hispidus collected near the residence of a human case in Dade County, Florida.
Other Hantaviruses
Several members of the hantavirus genus cause different forms of hemorrhagic fever with renal syndrome (HFRS), an ancient disease first described in Russia in 1913. The four viruses that are associated with HFRS, each named for the region from where they were first isolated, have different primary rodent hosts: Apodemus agrarius (the striped field mouse) for Hantaan virus, Rattus norvegicus (the Norway rat) and Rattus rattus (the black rat) for Seoul virus, Clethrionomys glareolus (the bank vole) for Puumala virus, and Apodemus flavicollis (the yellow-necked field mouse) for Dobrava virus. Hantaan virus from Korea and Dobrava virus from Slovenia are associated with a severe form of HFRS characterized by renal failure that can precede pulmonary edema and disseminated intravascular coagulation (DIC), with estimated mortality rates of 5% to 15%. A moderate form of HFRS caused by Seoul virus (which, along with its host, is distributed worldwide) is responsible for thousands of Eurasian cases annually. Serologic evidence for infection with Seoul-like hantaviruses has been found in rodents in major cities of the United States, and this virus was recently implicated in human cases of HFRS in Baltimore. One report has also associated Seoul virus with chronic renal disease. A mild form of HFRS, caused by Puumala virus, is responsible for nephropathia epidemica in Scandinavia, with an estimated mortality rate of 1% to 3%.
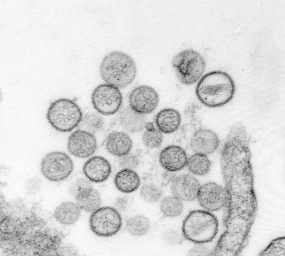
Thin-section electron micrograph of Sin Nombre virus isolate, a causative agent of hantavirus pulmonary syndrome (HPS). From the 1993 outbreak of HPS in the southwestern United States. Image courtesy of Cynthia Goldsmith, Sherif Zaki, and Luanne Elliott
| Hantaviruses | Geographic Region | Reservoir | Pathology | Mortality |
|---|---|---|---|---|
| Hantaan | Asia | Field mouse | Renal | 5-15% |
| Seoul | Worldwide | Domestic rat | Renal | 1% |
| Puumala | Northern Europe | Bank vole | Renal | 1% |
| Prospect Hill | United States | Meadow vole | No known human disease | N/A |
| Sin Nombre | North America | Deer mouse | Pulmonary | 50% |
| Feature | HFRS | HPS |
|---|---|---|
| Major target organ | Kidney | Lung |
| First phase | Febrile | Febrile “prodrome” |
| Second phase | Shock | Shock, pulmonary edema |
| Evolution | Oliguria, diureses, convalescence | Diureses, convalescence |
| Mortality | 1-15% | 50% |
Epidemiology in the Virology Laboratory
During the outbreak in 1993, definitive proof that the agent causing HPS was a novel hantavirus was obtained using a genetic detection assay. Oligonucleotide primers were designed on the basis of regions of the M segment (G2 coding region) conserved among hantaviruses and were used in a nested RT-PCR assay to amplify hantavirus-specific DNA fragments from RNA extracted from the tissues of patients. The amplified DNA fragments were then sequenced. Comparative and phylogenetic analyses of derived sequence data demonstrated that the hantavirus associated with the HPS outbreak (SNV) was a novel virus most closely related to Prospect Hill virus (PHV). In addition, a direct genetic link was made between the human HPS cases and the virus harbored byperidomestic P. maniculatus rodents. Characterization of hantaviral genetic sequences recovered from human tissues demonstrated that these sequences were identical to those from rodents captured at the site of the patient’s presumed infection. This characterization has continued to facilitate identification of the site of infection when more than one such site exists and therefore focus the public health response. These techniques also allow implication of a specific rodent host in areas of overlapping hosts.
Sin Nombre Virus Sequencing
The entire genomic sequence of SNV has subsequently been determined by using RNA extracted from autopsy material as well as RNA extracted from cell culture-adapted virus. The L RNA is 6562 nucleotides (nt) in length; the M RNA is 3696 nt long; and the S RNA is 2059 to 2060 nt long. Interestingly, when the prototype sequence (NMH10) of SNV detected in tissues from an HPS case was compared with the sequence of the SNV isolate (NMR11; isolated in Vero E6 cells from P. maniculatus trapped in the residence of the same case), only 16 nucleotide changes were found, and none of these changes resulted in alterations in amino acid sequences of viral proteins. It had been assumed that in the process of adaptation to cell culture, selection of SNV variants which grow optimally in cell culture would occur, and selected variants would differ genetically from the parental virus. Though NMH10 and NMR11 are identical in protein sequence, nucleotide substitutions in nontranslated regions of the genome could be responsible for altered viral phenotypes, as could changes in protein glycosylation or virus membrane components.
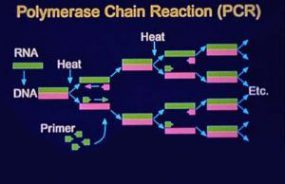
The nested RT-PCR assay developed during the initial HPS outbreak provided a rapid method for the genetic characterization of novel hantaviruses that did not require a virus isolate. Numerous new hantaviruses have been detected by RT-PCR in rodent tissues but have yet to be associated with human disease. These include El Moro Canyon virus associated with the western harvest mouse (Reithrodontomys megalotis), Tula virus with Microtus arvalis and M. rossiaemeridionalis, Rio Segundo virus with the Mexican harvest mouse (R. mexicanus), Isla Vista virus with the California vole (M. californicus), and Prospect Hill-like viruses in Microtus species.
Phylogenetic Analysis
Phylogenetic analysis of Old World and American hantaviruses indicates that the relationship among hantaviruses corresponds with the phylogeny of their rodent hosts. Viruses of rodents belonging to the subfamily Murinae are monophyletic as are hantaviruses of arvicoline and sigmodontine rodents, suggesting that long-term virus-rodent coevolution is taking place. Hantavirus evolution is best understood as co-evolution within specific lineages in the rodent family Muridae. The apparent coupling between hantaviruses and their rodent hosts suggests that viruses of sigmodontine rodents share a common ancestor, as do viruses of the subfamily Arvicolinae and Murinae. This coupling also has a geographic and clinical correlate: viruses found in Old World murine rodents, including Hantaan virus (HTNV), Seoul virus (SEOV) and Dobrava virus, are associated with HFRS in Eurasia. By contrast, viruses carried by New World sigmodontine rodents, including SNV Black Creek Canal virus (BCCV) and Bayou virus (BAYV), are associated with HPS in the Americas. This distinction can narrow the search for a rodent host for newly discovered HPS-like diseases and suggest disease implications for the various new viruses being genetically amplified from rodents.
Sequence Divergence
Detection and characterization of Sin Nombre-like viruses in P. maniculatus and P. leucopus populations have shown that multiple phylogenetic lineages of SNV exist in North America, and in some cases similar viruses are detected in both Peromyscus species. Sequence divergence among SNV genes has been shown to be as high as 23% nucleotide dissimilarity and 7% amino acid dissimilarity. Comparison of SNV sequences with those of other hantaviruses provides no obvious explanation as to why SNV and related viruses cause HPS while other hantaviruses are associated with HFRS. The development of a reverse genetics system for manipulation of virus genomes and an animal model for studying pathogenesis will be necessary to define the molecular mechanism(s) of SNV pathogenicity.
Genomic reassortment by RNA viruses with segmented genomes is well documented and has the potential to produce viruses with altered biological activity, host range, and disease potential. For example, segment reassortment among influenza virus strains (antigenic shift) is thought to be responsible for influenza pandemics. Genomic reassortment among SNV variants is known to occur in nature, but the precise role of genomic reassortment in the epidemiology of HPS and HFRS is unknown.