Persons using assistive technology might not be able to fully access information in this file. For assistance, please send e-mail to: mmwrq@cdc.gov. Type 508 Accommodation and the title of the report in the subject line of e-mail.
Guidelines for the Prevention and Treatment of Opportunistic Infections Among HIV-Exposed and HIV-Infected Children
Recommendations from CDC, the National Institutes of Health, the HIV Medicine Association of the Infectious Diseases Society of America, the Pediatric Infectious Diseases Society, and the American Academy of Pediatrics
Summary
This report updates and combines into one document earlier versions of guidelines for preventing and treating opportunistic infections (OIs) among HIV-exposed and HIV-infected children, last published in 2002 and 2004, respectively. These guidelines are intended for use by clinicians and other health-care workers providing medical care for HIV-exposed and HIV-infected children in the United States. The guidelines discuss opportunistic pathogens that occur in the United States and one that might be acquired during international travel (i.e., malaria). Topic areas covered for each OI include a brief description of the epidemiology, clinical presentation, and diagnosis of the OI in children; prevention of exposure; prevention of disease by chemoprophylaxis and/or vaccination; discontinuation of primary prophylaxis after immune reconstitution; treatment of disease; monitoring for adverse effects during treatment; management of treatment failure; prevention of disease recurrence; and discontinuation of secondary prophylaxis after immune reconstitution. A separate document about preventing and treating of OIs among HIV-infected adults and postpubertal adolescents (Guidelines for the Prevention and Treatment of Opportunistic Infections in HIV-Infected Adults and Adolescents) was prepared by a working group of adult HIV and infectious disease specialists.
The guidelines were developed by a panel of specialists in pediatric HIV infection and infectious diseases (the Pediatric Opportunistic Infections Working Group) from the U.S. government and academic institutions. For each OI, a pediatric specialist with content-matter expertise reviewed the literature for new information since the last guidelines were published; they then proposed revised recommendations at a meeting at the National Institutes of Health (NIH) in June 2007. After these presentations and discussions, the guidelines underwent further revision, with review and approval by the Working Group, and final endorsement by NIH, CDC, the HIV Medicine Association (HIVMA) of the Infectious Diseases Society of America (IDSA), the Pediatric Infectious Disease Society (PIDS), and the American Academy of Pediatrics (AAP). The recommendations are rated by a letter that indicates the strength of the recommendation and a Roman numeral that indicates the quality of the evidence supporting the recommendation so readers can ascertain how best to apply the recommendations in their practice environments.
An important mode of acquisition of OIs, as well as HIV infection among children, is from their infected mother; HIV-infected women coinfected with opportunistic pathogens might be more likely than women without HIV infection to transmit these infections to their infants. In addition, HIV-infected women or HIV-infected family members coinfected with certain opportunistic pathogens might be more likely to transmit these infections horizontally to their children, resulting in increased likelihood of primary acquisition of such infections in the young child. Therefore, infections with opportunistic pathogens might affect not just HIV-infected infants but also HIV-exposed but uninfected infants who become infected by the pathogen because of transmission from HIV-infected mothers or family members with coinfections. These guidelines for treating OIs in children therefore consider treatment of infections among all children, both HIV-infected and uninfected, born to HIV-infected women.
Additionally, HIV infection is increasingly seen among adolescents with perinatal infection now surviving into their teens and among youth with behaviorally acquired HIV infection. Although guidelines for postpubertal adolescents can be found in the adult OI guidelines, drug pharmacokinetics and response to treatment may differ for younger prepubertal or pubertal adolescents. Therefore, these guidelines also apply to treatment of HIV-infected youth who have not yet completed pubertal development.
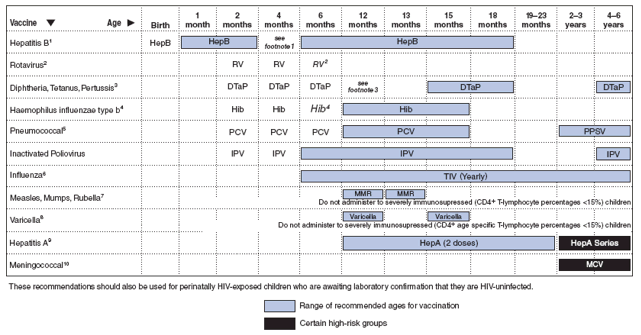
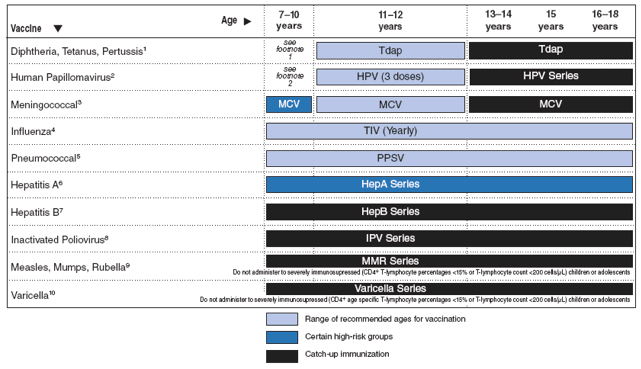
Major changes in the guidelines include 1) greater emphasis on the importance of antiretroviral therapy for preventing and treating OIs, especially those OIs for which no specific therapy exists; 2) information about the diagnosis and management of immune reconstitution inflammatory syndromes; 3) information about managing antiretroviral therapy in children with OIs, including potential drug--drug interactions; 4) new guidance on diagnosing of HIV infection and presumptively excluding HIV infection in infants that affect the need for initiation of prophylaxis to prevent Pneumocystis jirovecii pneumonia (PCP) in neonates; 5) updated immunization recommendations for HIV-exposed and HIV-infected children, including hepatitis A, human papillomavirus, meningococcal, and rotavirus vaccines; 6) addition of sections on aspergillosis; bartonella; human herpes virus-6, -7, and -8; malaria; and progressive multifocal leukodystrophy (PML); and 7) new recommendations on discontinuation of OI prophylaxis after immune reconstitution in children. The report includes six tables pertinent to preventing and treating OIs in children and two figures describing immunization recommendations for children aged 0--6 years and 7--18 years.
Because treatment of OIs is an evolving science, and availability of new agents or clinical data on existing agents might change therapeutic options and preferences, these recommendations will be periodically updated and will be available at http://AIDSInfo.nih.gov.
Background
Opportunistic Infections in HIV-Infected Children in the Era of Potent Antiretroviral Therapy
In the pre-antiretroviral era and before development of potent combination highly active antiretroviral treatment (HAART) regimens, opportunistic infections (OIs) were the primary cause of death in human immunodeficiency virus (HIV)-infected children (1). Current HAART regimens suppress viral replication, provide significant immune reconstitution, and have resulted in a substantial and dramatic decrease in acquired immunodeficiency syndrome (AIDS)-related OIs and deaths in both adults and children (2--4). In an observational study from pediatric clinical trial sites in the United States, Pediatric AIDS Clinical Trials Group (PACTG) 219, the incidence of the most common initial OIs in children during the potent HAART era (study period 2000--2004) was substantially lower than the incidence in children followed at the same sites during the pre-HAART era (study period 1988--1998) (1,3). For example, the incidence for bacterial pneumonia decreased from 11.1 per 100 child-years during the pre-HAART era to 2.2 during the HAART era; bacteremia from 3.3 to 0.4 per 100 child-years; herpes zoster from 2.9 to 1.1 per 100 child-years; disseminated Mycobacterium avium complex (MAC) from 1.8 to 0.14 per 100 child-years; and Pneumocystis jirovecii pneumonia (PCP) from 1.3 to 0.09 per 100 child-years.
Despite this progress, prevention and management of OIs remain critical components of care for HIV-infected children. OIs continue to be the presenting symptom of HIV infection among children whose HIV-exposure status is not known (e.g., because of lack of maternal antenatal HIV testing). For children with known HIV infection, barriers such as parental substance abuse may limit links to appropriate care where indications for prophylaxis would be evaluated. HIV-infected children eligible for primary or secondary OI prophylaxis might fail to be treated because they are receiving suboptimal medical care. Additionally, adherence to multiple drugs (antiretroviral drugs and concomitant OI prophylactic drugs) may prove difficult for the child or family. Multiple drug-drug interactions of OI, antiretroviral, and other drugs resulting in increased adverse events and decreased treatment efficacy may limit the choice and continuation of both HAART and prophylactic regimens. OIs continue to occur in children in whom drug resistance causes virologic and immunologic failure. In PACTG 219, lack of a sustained response to HAART predicted OIs in children (5). Finally, immune reconstitution inflammatory syndrome (IRIS), initially described in HIV-infected adults but also seen in HIV-infected children, can complicate treatment of OIs when HAART is started or when optimization of a failing regimen is attempted in a patient with acute OI. Thus, preventing and treating OIs in HIV-infected children remains important even in an era of potent HAART.
History of the Guidelines
In 1995, the U.S. Public Health Service and the Infectious Diseases Society of America (IDSA) developed guidelines for preventing OIs among adults, adolescents, and children infected with HIV (6). These guidelines, developed for health-care providers and their HIV-infected patients, were revised in 1997, 1999, and 2002 (7,8). In 2001, the National Institutes of Health, IDSA, and CDC convened a working group to develop guidelines for treating HIV-associated OIs, with a goal of providing evidence-based guidelines on treatment and prophylaxis. In recognition of unique considerations for HIV-infected infants, children, and adolescents---including differences between adults and children in mode of acquisition, natural history, diagnosis, and treatment of HIV-related OIs---a separate pediatric OI guidelines writing group was established. The pediatric OI treatment guidelines were initially published in December 2004 (9).
The current document combines recommendations for preventing and treating OIs in HIV-exposed and HIV-infected children into one document; it accompanies a similar document on preventing and treating OIs among HIV-infected adults prepared by a separate group of adult HIV and infectious disease specialists. Both sets of guidelines were prepared by the Opportunistic Infections Working Group under the auspices of the Office of AIDS Research (OAR) of the National Institutes of Health. Pediatric specialists with expertise in specific OIs reviewed the literature since the last publication of the prevention and treatment guidelines, conferred over several months, and produced draft guidelines. The Pediatric OI Working Group reviewed and discussed recommendations at a meeting in Bethesda, Maryland, on June 25--26, 2007. After the meeting, the document was revised, then reviewed and electronically approved by the writing group members. The final report was further reviewed by the core Writing Group, the Office of AIDS Research, experts at CDC, the HIV Medicine Association of IDSA, the Pediatric Infectious Diseases Society, and the American Academy of Pediatrics before final approval and publication.
Why Pediatric Prevention and Treatment Guidelines?
Mother-to-child transmission is an important mode of acquisition of OIs and HIV infection in children. HIV-infected women coinfected with opportunistic pathogens might be more likely than women without HIV infection to transmit these infections to their infants. For example, greater rates of perinatal transmission of hepatitis C and cytomegalovirus (CMV) have been reported from HIV-infected than HIV-uninfected women (10,11). In addition, HIV-infected women or HIV-infected family members coinfected with certain opportunistic pathogens might be more likely to transmit these infections horizontally to their children, increasing the likelihood of primary acquisition of such infections in the young child. For example, Mycobacterium tuberculosis infection among children primarily reflects acquisition from family members who have active tuberculosis (TB) disease, and increased incidence and prevalence of TB among HIV-infected persons is well documented. HIV-exposed or -infected children in the United States might have a higher risk for exposure to M. tuberculosis than would comparably aged children in the general U.S. population because of residence in households with HIV-infected adults (12). Therefore, OIs might affect not only HIV-infected infants but also HIV-exposed but uninfected infants who become infected with opportunistic pathogens because of transmission from HIV-infected mothers or family members with coinfections. Guidelines for treating OIs in children must consider treatment of infections among all children---both HIV-infected and HIV-uninfected---born to HIV-infected women.
The natural history of OIs among children might differ from that among HIV-infected adults. Many OIs in adults are secondary to reactivation of opportunistic pathogens, which often were acquired before HIV infection when host immunity was intact. However, OIs among HIV-infected children more often reflect primary infection with the pathogen. In addition, among children with perinatal HIV infection, the primary infection with the opportunistic pathogen occurs after HIV infection is established and the child's immune system already might be compromised. This can lead to different manifestations of specific OIs in children than in adults. For example, young children with TB are more likely than adults to have nonpulmonic and disseminated infection, even without concurrent HIV infection.
Multiple difficulties exist in making laboratory diagnoses of various infections in children. A child's inability to describe the symptoms of disease often makes diagnosis more difficult. For infections for which diagnosis is made by laboratory detection of specific antibodies (e.g., the hepatitis viruses and CMV), transplacental transfer of maternal antibodies that can persist in the infant for up to 18 months complicates the ability to make a diagnosis in young infants. Assays capable of directly detecting the pathogen are required to diagnose such infections definitively in infants. In addition, diagnosing the etiology of lung infections in children can be difficult because children usually do not produce sputum, and more invasive procedures, such as bronchoscopy or lung biopsy, might be needed to make a more definitive diagnosis.
Data related to the efficacy of various therapies for OIs in adults usually can be extrapolated to children, but issues related to drug pharmacokinetics, formulation, ease of administration, and dosing and toxicity require special considerations for children. Young children in particular metabolize drugs differently from adults and older children, and the volume of distribution differs. Unfortunately, data often are lacking on appropriate drug dosing recommendations for children aged <2 years.
The prevalence of different opportunistic pathogens among HIV-infected children during the pre-HAART era varied by child age, previous OI, immunologic status, and pathogen (1). During the pre-HAART era, the most common OIs among children in the United States (event rates >1.0 per 100 child-years) were serious bacterial infections (most commonly pneumonia, often presumptively diagnosed, and bacteremia), herpes zoster, disseminated MAC, PCP, and candidiasis (esophageal and tracheobronchial disease). Less commonly observed OIs (event rate <1.0 per 100 child-years) included CMV disease, cryptosporidiosis, TB, systemic fungal infections, and toxoplasmosis (3,4). History of a previous AIDS-defining OI predicted development of a new infection. Although most infections occurred among substantially immunocompromised children, serious bacterial infections, herpes zoster, and TB occurred across the spectrum of immune status.
Descriptions of pediatric OIs in children receiving HAART have been limited. As with HIV-infected adults, substantial decreases in mortality and morbidity, including OIs, have been observed among children receiving HAART (2). Although the number of OIs has substantially decreased during the HAART era, HIV-associated OIs and other related infections continue to occur among HIV-infected children (3,13).
In contrast to recurrent serious bacterial infections, some of the protozoan, fungal, or viral OIs complicating HIV are not curable with available treatments. Sustained, effective HAART, resulting in improved immune status, has been established as the most important factor in controlling OIs among both HIV-infected adults and children (14). For many OIs, after treatment of the initial infectious episode, secondary prophylaxis in the form of suppressive therapy is indicated to prevent recurrent clinical disease from reactivation or reinfection (15).
These guidelines are a companion to the Guidelines for Prevention and Treatment of Opportunistic Infections in HIV-Infected Adults and Adolescents (16). Treatment of OIs is an evolving science, and availability of new agents or clinical data on existing agents might change therapeutic options and preferences. As a result, these recommendations will need to be periodically updated.
Because the guidelines target HIV-exposed and -infected children in the United States, the opportunistic pathogens discussed are those common to the United States and do not include certain pathogens (e.g., Penicillium marneffei) that might be seen more frequently in resource-limited countries or that are common but seldom cause chronic infection (e.g., chronic parvovirus B19 infection). The document is organized to provide information about the epidemiology, clinical presentation, diagnosis, and treatment for each pathogen. The most critical treatment recommendation is accompanied by a rating that includes a letter and a roman numeral and is similar to the rating systems used in other U.S. Public Health Service/Infectious Diseases Society of America guidelines (17). Recommendations unrelated to treatment were not graded, with some exceptions. The letter indicates the strength of the recommendation, which is based on the opinion of the Working Group, and the roman numeral reflects the nature of the evidence supporting the recommendation (Box 1). Because licensure of drugs for children often relies on efficacy data from adult trials and safety data in children, recommendations sometimes may need to rely on data from clinical trials or studies in adults.
Tables at the end of this document summarize recommendations for preventing OIs in children (Tables 1--3); treatment of OIs in children (Table 4); drug preparation and toxicity information for children (Table 5); drug-drug interactions (Table 6), and vaccination recommendations for HIV-infected children and adolescents (Figures 1 and 2).
Diagnosis of HIV Infection and Presumptive Lack of HIV Infection in Children with Perinatal HIV Exposure
Because maternal antibody persists in children up to 18 months of age, virologic tests (usually HIV DNA or RNA assays) are needed to determine infection status in children aged <18 months. The CDC surveillance definition states a child is considered definitively infected if he or she has positive virologic results on two separate specimens or is aged >18 months and has either a positive virologic test or a positive confirmed HIV-antibody test.
CDC has revised laboratory criteria to allow presumptive exclusion of HIV infection at an earlier age for surveillance (Box 2) (http://www.cdc.gov/mmwr/preview/mmwrhtml/rr5710a1.htm). A child who has not been breast-fed is presumed to be uninfected if he or she has no clinical or laboratory evidence of HIV infection and has two negative virologic tests both obtained at ≥2 weeks of age and one obtained at ≥4 weeks of age and no positive viralogic tests; or one negative virologic test at ≥8 weeks of age and no positive virologic tests; or one negative HIV-antibody test at ≥6 months of age. Definitive lack of infection is confirmed by two negative viral tests, both of which were obtained at ≥1 month of age and one of which was obtained at ≥4 months of age, or at least two negative HIV-antibody tests from separate specimens obtained at ≥6 months of age. The new presumptive definition of "uninfected" may allow clinicians to avoid starting PCP prophylaxis in some HIV-exposed infants at age 6 weeks (see PCP section).
Antiretroviral Therapy and Management of Opportunistic Infections
Studies in adults and children have demonstrated that HAART reduces the incidence of OIs and improves survival, independent of the use of OI antimicrobial prophylaxis. HAART can improve or resolve certain OIs, such as cryptosporidiosis or microsporidiosis infection, for which effective specific treatments are not available. However, potent HAART does not replace the need for OI prophylaxis in children with severe immune suppression. Additionally, initiation of HAART in persons with an acute or latent OI can lead to IRIS, an exaggerated inflammatory reaction that can clinically worsen disease and require use of anti-inflammatory drugs (see IRIS section below).
Specific data are limited to guide recommendations for when to start HAART in children with an acute OI and how to manage HAART when an acute OI occurs in a child already receiving HAART. The decision of when to start HAART in a child with an acute or latent OI needs to be individualized and will vary by the degree of immunologic suppression in the child before he or she starts HAART. Similarly, in a child already receiving HAART who develops an OI, management will need to account for the child's clinical, viral, and immune status on HAART and the potential drug-drug interactions between HAART and the required OI drug regimen.
Immune Reconstitution Inflammatory Syndrome
As in adults, antiretroviral therapy improves immune function and CD4 cell count in HIV-infected children; within the first few months after starting treatment, HIV viral load sharply decreases and the CD4 count rapidly increases. This results in increased capacity to mount inflammatory reactions. After initiation of HAART, some patients develop a paradoxical inflammatory response by their reconstituted immune system to infectious or noninfectious antigens, resulting in apparent clinical worsening. This is referred to as IRIS, and although primarily reported in adults initiating therapy, it also has been reported in children (18--28).
IRIS can occur after initiation of HAART because of worsening of an existing active, latent, or occult OI, where infectious pathogens previously not recognized by the immune system now evoke an immune response. This inflammatory response often is exaggerated in comparison with the response in patients who have normal immune systems (referred to by some experts as immune reconstitution disease). An example is activation of latent or occult TB after initiation of antiretroviral therapy (referred to by some experts as "unmasking IRIS"). Alternatively, clinical recrudescence of a successfully treated infection can occur, with paradoxical, symptomatic relapse despite microbiologic treatment success and sterile cultures (referred to as "paradoxical IRIS"). In this case, reconstitution of antigen-specific T-cell--mediated immunity occurs with activation of the immune system after initiation of HAART against persisting antigens, whether present as dead, intact organisms or as debris.
The pathologic process of IRIS is inflammatory and not microbiologic in etiology. Thus, distinguishing IRIS from treatment failure, antimicrobial resistance, or noncompliance is important. In therapeutic failure, a microbiologic culture should reveal the continued presence of an infectious organism, whereas in paradoxical IRIS, follow-up cultures are most often sterile. However, with "unmasking" IRIS, viable pathogens may be isolated.
IRIS is described primarily on the basis of reports of cases in adults. A proposed clinical definition is worsening symptoms of inflammation or infection temporally related to starting HAART that are not explained by newly acquired infection or disease, the usual course of a previously acquired disease, or HAART toxicity in a patient with ≥1 log10 decrease in plasma HIV RNA (29).
The timing of IRIS after initiation of HAART in adults has varied, with most cases occurring during the first 2--3 months after initiation; however, as many as 30% of IRIS cases can present beyond the first 3 months of treatment. Later-onset IRIS may result from an immune reaction against persistent noninfectious antigen. The onset of antigen clearance varies, but antigen or antigen debris might persist long after microbiologic sterility. For example, after pneumococcal bacteremia, the C-polysaccharide antigen can be identified in the urine of 40% of HIV-infected adults 1 month after successful treatment; similarly, mycobacterial DNA can persist several months past culture viability.
In adults, IRIS most frequently has been observed after initiation of therapy in persons with mycobacterial infections (including MAC and M. tuberculosis), PCP, cryptococcal infection, CMV, varicella zoster or herpes virus infections, hepatitis B and C infections, toxoplasmosis, and progressive multifocal leukoencephalopathy (PML). Reactions also have been described in children who had received bacille Calmette-Guérin (BCG) vaccine and later initiated HAART (22,25,26,28). In a study of 153 symptomatic children with CD4 <15% at initiation of therapy in Thailand, the incidence of IRIS was 19%, with a median time of onset of 4 weeks after start of HAART; children who developed IRIS had lower baseline CD4 percentage than did children who did not develop IRIS (24).
No randomized controlled trials have been published evaluating treatment of IRIS. Treatment has been based on severity of disease (CIII). For mild cases, observation alone with close clinical and laboratory monitoring may be sufficient. For moderate cases, nonsteroidal anti-inflammatory drugs have been used to ameliorate symptoms. For severe cases, corticosteroids, such as dexamethasone, have been used. However, the optimal dosing and duration of therapy are unknown, and inflammation can take weeks to months to subside. During this time, HAART should be continued.
Initiation of HAART for an Acute OI in Treatment-Naïve Children
The ideal time to initiate HAART for an acute OI is unknown. The benefit of initiating HAART is improved immune function, which could result in faster resolution of the OI. This is particularly important for OIs for which effective therapeutic options are limited or not available, such as for cryptosporidiosis, microsporidiosis, PML, and Kaposi sarcoma (KS). However, potential problems exist when HAART and treatment for the OI are initiated simultaneously. These include drug-drug interactions between the antiretroviral and antimicrobial drugs, particularly given the limited repertoire of antiretroviral drugs available for children than for adults; issues related to toxicity, including potential additive toxicity of antiretroviral and OI drugs and difficulty in distinguishing HAART toxicity from OI treatment toxicity; and the potential for IRIS to complicate OI management.
The primary consideration in delaying HAART until after initial treatment of the acute OI is risk for death during the delay. Although the short-term risk for death in the United States during a 2-month HAART delay may be relatively low, mortality in resource-limited countries is significant. IRIS is more likely to occur in persons with advanced HIV infection and higher OI-specific antigenic burdens, such as those who have disseminated infections or a shorter time from an acute OI onset to start of HAART. However, in the absence of an OI with central nervous system (CNS) involvement, such as cryptococcal meningitis, most IRIS events, while potentially resulting in significant morbidity, do not result in death. With CNS IRIS or in resource-limited countries, significant IRIS-related death may occur with simultaneous initiation of HAART and OI treatment; however, significant mortality also occurs in the absence of HAART.
Because no randomized trials exist in either adults or children to address the optimal time for starting HAART when an acute OI is present, decisions need to be individualized for each child. The timing is a complex decision based on the severity of HIV disease, efficacy of standard OI-specific treatment, social support system, medical resource availability, potential drug-drug interactions, and risk for IRIS. Most experts believe that for children who have OIs that lack effective treatment (e.g., cryptosporidiosis, microsporidiosis, PML, KS), the early benefit of potent HAART outweighs any increased risk, and potent HAART should begin as soon as possible (AIII). For other OIs, such as TB, MAC, PCP, and cryptococcal meningitis, awaiting a response to therapy may be warranted before initiating HAART (CIII).
Management of Acute OIs in HIV-Infected Children Receiving HAART
OIs in HIV-infected children soon after initiation of HAART (within 12 weeks) may be subclinical infections unmasked by HAART-related improvement in immune function, also known as "unmasking IRIS" and occurring usually in children who have more severe immune suppression at initiation of HAART. This does not represent a failure of HAART but rather a sign of immune reconstitution (see IRIS section). In such situations, HAART should be continued and treatment for the OI initiated (AIII). Assessing the potential for drug-drug interactions between the antiretroviral and antimicrobial drugs and whether treatment modifications need to be made is important.
In children who develop an OI after receiving >12 weeks of HAART with virologic and immunologic response to therapy, it can be difficult to distinguish between later-onset IRIS (such as a "paradoxical IRIS" reaction where the reconstituted immune system demonstrates an inflammatory reaction to a noninfectious antigen) and incomplete immune reconstitution with HAART allowing occurrence of a new OI. In such situations, HAART should be continued, and if microbiologic evaluation demonstrates organisms by stain or culture, specific OI-related therapy should be initiated (AII).
OIs also can occur in HIV-infected children experiencing virologic and immunologic failure on HAART and represent clinical failure of therapy. In this situation, treatment of the OI should be initiated, viral resistance testing performed, and the child's HAART regimen reassessed, as described in pediatric antiretroviral guidelines (14).
Preventing Vaccine-Preventable Diseases in HIV-Infected Children and Adolescents
Vaccines are an extremely effective primary prevention tool, and vaccines that protect against 16 diseases are recommended for routine use in children and adolescents in the United States. Vaccination schedules for children aged 0--6 years and 7--18 years are published annually (http://www.cdc.gov/vaccines/recs/schedules/default.htm). These schedules are compiled from approved vaccine-specific policy recommendations and are standardized among the major vaccine policy-setting and vaccine-delivery organizations (e.g., Advisory Committee on Immunization Practices [ACIP], American Academy of Pediatrics, American Association of Family Physicians).
HIV-infected children should be protected from vaccine-preventable diseases. Most vaccines recommended for routine use can be administered safely to HIV-exposed or HIV-infected children. The recommended vaccination schedules for 2009 for HIV-exposed and HIV-infected children aged 0--6 years and 7--18 years were approved by the ACIP through October 2008 (Figures 1 and 2). These schedules will be updated periodically to reflect additional ACIP-approved vaccine recommendations that pertain to HIV-exposed or HIV-infected children.
All inactivated vaccines can be administered safely to persons with altered immunocompetence whether the vaccine is a killed whole organism or a recombinant, subunit, toxoid, polysaccharide, or polysaccharide protein-conjugate vaccine. If inactivated vaccines are indicated for persons with altered immunocompetence, the usual doses and schedules are recommended. However, the effectiveness of such vaccinations might be suboptimal (30).
Persons with severe cell-mediated immune deficiency should not receive live attenuated vaccines. However, children with HIV infection are at higher risk than immunocompetent children for complications of varicella, herpes zoster, and measles. On the basis of limited safety, immunogenicity, and efficacy data among HIV-infected children, varicella and measles-mumps-rubella vaccines can be considered for HIV-infected children who are not severely immunosuppressed (i.e., those with age-specific CD4 cell percentages of ≥15%) (30--32). Practitioners should consider the potential risks and benefits of administering rotavirus vaccine to infants with known or suspected altered immunocompetence; consultation with an immunologist or infectious diseases specialist is advised. There are no safety or efficacy data related to the administration of rotavirus vaccine to infants who are potentially immunocompromised, including those who are HIV-infected (33). However, two considerations support vaccination of HIV-exposed or -infected infants: first, the HIV diagnosis may not be established in infants born to HIV-infected mothers before the age of the first rotavirus vaccine dose (only 1.5%--3.0% of HIV-exposed infants in the United States will be determined to be HIV-infected); and second, vaccine strains of rotavirus are considerably attenuated.
Consult the specific ACIP statements (available at http://www.cdc.gov/vaccines/pubs/ACIP-list.htm) for more detail regarding recommendations, precautions, and contraindications for use of specific vaccines (http://www.cdc.gov/mmwr/PDF/rr/rr4608.pdf and http://www.cdc.gov/mmwr/pdf/rr/rr5602.pdf) (31--44).
Bacterial Infections
Bacterial Infections, Serious and Recurrent
Epidemiology
During the pre-HAART era, serious bacterial infections were the most commonly diagnosed OIs in HIV-infected children, with an event rate of 15 per 100 child-years (1). Pneumonia was the most common bacterial infection (11 per 100 child-years), followed by bacteremia (3 per 100 child-years), and urinary tract infection (2 per 100 child-years). Other serious bacterial infections, including osteomyelitis, meningitis, abscess, and septic arthritis, occurred at rates <0.2 per 100 child-years. More minor bacterial infections such as otitis media and sinusitis were particularly common (17--85 per 100 child-years) in untreated HIV-infected children (45).
With the advent of HAART, the rate of pneumonia has decreased to 2.2--3.1 per 100 child-years (3,46), similar to the rate of 3--4 per 100 child-years in HIV-uninfected children (47,48). The rate of bacteremia/sepsis during the HAART era also has decreased dramatically to 0.35--0.37 per 100 child-years (3,4,46), but this rate remains substantially higher than the rate of <0.01 per 100 child-years in HIV-uninfected children (49,50). Sinusitis and otitis rates among HAART-treated children are substantially lower (2.9--3.5 per 100 child-years) but remain higher than rates in children who do not have HIV infection (46).
Acute pneumonia, often presumptively diagnosed in children, was associated with increased risk for long-term mortality among HIV-infected children in one study during the pre-HAART era (51). HIV-infected children with pneumonia are more likely to be bacteremic and to die than are HIV-uninfected children with pneumonia (52). Chronic lung disease might predispose persons to development of acute pneumonia; in one study, the incidence of acute lower respiratory tract infection in HIV-infected children with chronic lymphoid interstitial pneumonitis was approximately 10-fold higher than in a community-based study of HIV-uninfected children (53). Chronically abnormal airways probably are more susceptible to infectious exacerbations (similar to those in children and adults with bronchiectasis or cystic fibrosis) caused by typical respiratory bacteria (Streptococcus pneumoniae, nontypeable Haemophilus influenzae) and Pseudomonas spp.
S. pneumoniae was the most prominent invasive bacterial pathogen in HIV-infected children both in the United States and worldwide, accounting for >50% of bacterial bloodstream infections in HIV-infected children (1,4,54--57). HIV-infected children have a markedly higher risk for pneumococcal infection than do HIV-uninfected children (58,59). In the absence of HAART, the incidence of invasive pneumococcal disease was 6.1 per 100 child-years among HIV-infected children through age 7 years (60), whereas among children treated with HAART, the rate of invasive pneumococcal disease decreased by about half, to 3.3 per 100 child-years (46). This is consistent with the halving of invasive pneumococcal disease rates in HIV-infected adults receiving HAART compared with rates in those not receiving HAART (61). Among children with invasive pneumococcal infections, study results vary on whether penicillin-resistant pneumococcal strains are more commonly isolated from HIV-infected than HIV-uninfected persons (56,60,62--64). Reports among children without HIV infection have not demonstrated a difference in the case-fatality rate between those with penicillin-susceptible and those with nonsusceptible pneumococcal infections (case-fatality rate was associated with severity of disease and underlying illness) (65). Invasive disease caused by penicillin-nonsusceptible pneumococcus was associated with longer fever and hospitalization but not with greater risk for complications or poorer outcome in a study of HIV-uninfected children (66). Since routine use of seven-valent pneumococcal conjugate vaccine (PCV) in 2000, the overall incidence of drug-resistant pneumococcal infections has stabilized or decreased.
H. influenzae type b (Hib) also has been reported to have been more common in HIV-infected children before the availability of Hib vaccine. In a study in South African children who had not received Hib conjugate vaccine, the estimated relative annual rate of overall invasive Hib disease in children aged <1 year was 5.9 times greater among HIV-infected than HIV-uninfected children, and HIV-infected children were at greater risk for bacteremic pneumonia (67). However, Hib is unlikely to occur in HIV-infected children in most U.S. communities, where high rates of Hib vaccination result in very low rates of Hib nasopharyngeal colonization among contacts.
HIV-related immune dysfunction may increase the risk for invasive meningococcal disease in HIV-infected patients, but few cases have been reported (68--72). In a population-based study of invasive meningococcal disease in Atlanta, Georgia (72), as expected, the annual rate of disease was higher for 18- to 24-year-olds (1.17 per 100,000) than for all adults (0.5 per 100,000), but the estimated annual rate for HIV-infected adults was substantially higher (11.2 per 100,000). Risk for invasive meningococcal disease may be higher in HIV-infected adults. Specific data are not available on risk for meningococcal disease in younger HIV-infected children.
Although the frequency of gram-negative bacteremia is lower than that of gram-positive bacteremia among HIV-infected children, gram-negative bacteremia is more common among children with advanced HIV disease or immunosuppression and among children with central venous catheters. However, in children aged <5 years, gram-negative bacteremia also was observed among children with milder levels of immune suppression. In a study of 680 HIV-infected children in Miami, Florida, through 1997, a total of 72 (10.6%) had 95 episodes of gram-negative bacteremia; the predominant organisms identified in those with gram-negative bacteremia were P. aeruginosa (26%), nontyphoidal Salmonella (15%), Escherichia coli (15%), and H. influenzae (13%) (73). The relative frequency of the organisms varied over time, with the relative frequency of P. aeruginosa bacteremia increasing from 13% before 1984 to 56% during 1995--1997, and of Salmonella from 7% before 1984 to 22% during 1995--1997. However, H. influenzae was not observed after 1990 (presumably decreasing after incorporation of Hib vaccine into routine childhood vaccinations). The overall case-fatality rate for children with gram-negative bacteremia was 43%. Among Kenyan children with bacteremia, HIV infection increased the risk for nontyphoidal Salmonella and E. coli infections (74).
The presence of a central venous catheter increases the risk for bacterial infections in HIV-infected children, and the incidence is similar to that for children with cancer. The most commonly isolated pathogens in catheter-associated bacteremia in HIV-infected children are similar to those in HIV-negative children with indwelling catheters, including coagulase-negative staphylococci, S. aureus, enterococci, P. aeruginosa, gram-negative enteric bacilli, Bacillus cereus, and Candida spp. (57,75).
Data conflict about whether infectious morbidity increases in children who have been exposed to but not infected with HIV. In studies in developing countries, uninfected infants of HIV-infected mothers had higher mortality (primarily because of bacterial pneumonia and sepsis) than did those born to uninfected mothers (76,77). Advanced maternal HIV infection was associated with increased risk for infant death (76,77). In a study in Latin America and the Caribbean, 60% of 462 uninfected infants of HIV-infected mothers experienced infectious disease morbidity during the first 6 months of life, with the rate of neonatal infections (particularly sepsis) and respiratory infections higher than rates in comparable community-based studies (78). Among other factors, infections in uninfected infants were associated with more advanced maternal HIV disease and maternal smoking during pregnancy. However, in a study from the United States, the rate of lower respiratory tract infections in HIV-exposed, uninfected children was within the range reported for healthy children during the first year of life (79). In a separate study, the rate of overall morbidity (including but not specific to infections) decreased from 1990 through 1999 in HIV-exposed, uninfected children (80), although rates were not compared with an HIV-unexposed or community-based cohort.
Clinical Manifestations
Clinical presentation depends on the particular type of bacterial infection (e.g., bacteremia/sepsis, osteomyelitis/septic arthritis, pneumonia, meningitis, and sinusitis/otitis media) (81). HIV-infected children with invasive bacterial infections typically have a clinical presentation similar to children without HIV infection, with acute presentation and fever (59,60,82). HIV-infected children might be less likely than children without HIV infection to have leukocytosis (60).
The classical signs, symptoms, and laboratory test abnormalities that usually indicate invasive bacterial infection (e.g., fever and elevated white blood cell count) are usually present but might be lacking among HIV-infected children who have reduced immune competence (59,81). One-third of HIV-infected children not receiving HAART who have acute pneumonia have recurrent episodes (51). Resulting lung damage before initiation of HAART can lead to continued recurrent pulmonary infections, even in the presence of effective HAART.
In studies in Malawian and South African children with acute bacterial meningitis, the clinical presentations of children with and without HIV infection were similar (83,84). However, in the Malawi study, HIV-infected children were 6.4-fold more likely to have repeated episodes of meningitis than were children without HIV infection, although the study did not differentiate recrudescence from new infections (83). In both studies, HIV-infected children were more likely to die from meningitis than were children without HIV infection.
Diagnosis
Attempted isolation of a pathogenic organism from normally sterile sites (e.g., blood, cerebrospinal fluid [CSF], and pleural fluid) is strongly recommended. This is particularly important because of an increasing incidence of antimicrobial resistance, including penicillin-resistant S. pneumoniae and community-acquired methicillin-resistant S. aureus (MRSA).
Because of difficulties obtaining appropriate specimens (e.g., sputum) from young children, bacterial pneumonia is most often a presumptive diagnosis in a child with fever, pulmonary symptoms, and an abnormal chest radiograph unless an accompanying bacteremia exists. In the absence of a laboratory isolate, differentiating viral from bacterial pneumonia using clinical criteria can be difficult (85). In a study of intravenous immune globulin (IVIG) prophylaxis of bacterial infections, only a bacterial pathogen was identified in 12% of acute presumed bacterial pneumonia episodes (51). TB and PCP must always be considered in HIV-infected children with pneumonia. Presence of wheezing makes acute bacterial pneumonia less likely than other causes, such as viral pathogens, asthma exacerbation, "atypical" bacterial pathogens such as Mycoplasma pneumoniae, or aspiration. Sputum induction obtained by nebulization with hypertonic (5%) saline was evaluated for diagnosis of pneumonia in 210 South African infants and children (median age: 6 months), 66% of whom had HIV infection (86). The procedure was well-tolerated, and identified an etiology in 63% of children with pneumonia (identification of bacteria in 101, M. tuberculosis in 19, and PCP in 12 children). Blood and, if present, fluid from pleural effusion should be cultured.
Among children with bacteremia, a source for the bacteremia should be sought. In addition to routine chest radiographs, other diagnostic radiologic evaluations (e.g., abdomen, ultrasound studies) might be necessary among HIV-infected children with compromised immune systems to identify less apparent foci of infection (e.g., bronchiectasis, internal organ abscesses) (87--89). Among children with central venous catheters, both a peripheral and catheter blood culture should be obtained; if the catheter is removed, the catheter tip should be sent for culture. Assays for detection of bacterial antigens or evidence by molecular biology techniques are important for the diagnostic evaluation of HIV-infected children in whom unusual pathogens might be involved or difficult to identify or culture by standard techniques. For example, Bordetella pertussis and Chlamydia pneumoniae can be identified by a polymerase chain reaction (PCR) assay of nasopharyngeal secretions (85).
Prevention Recommendations
Preventing Exposure
Because S. pneumoniae and H. influenzae are common in the community, no effective way exists to eliminate exposure to these bacteria. However, routine use of conjugated seven-valent PCV and Hib vaccine in U.S. infants and young children has dramatically reduced vaccine type invasive disease and nasopharyngeal colonization, conferring herd protection of HIV-infected contacts because of decreased exposure to Hib and pneumoccal serotypes included in the vaccine.
Food. To reduce the risk for exposure to potential gastrointestinal (GI) bacterial pathogens, health-care providers should advise that HIV-infected children avoid eating the following raw or undercooked foods (including other foods that contain them): eggs, poultry, meat, seafood (especially raw shellfish), and raw seed sprouts. Unpasteurized dairy products and unpasteurized fruit juices also should be avoided. Of particular concern to HIV-infected infants and children is the potential for caretakers to handle these raw foods (e.g., during meal preparation) and then unknowingly transfer bacteria from their hands to the child's food, milk or formula or directly to the child. Hands, cutting boards, counters, and knives and other utensils should be washed thoroughly after contact with uncooked foods. Produce should be washed thoroughly before being eaten.
Pets. When obtaining a new pet, caregivers should avoid dogs or cats aged <6 months or stray animals. HIV-infected children and adults should avoid contact with any animals that have diarrhea and should wash their hands after handling pets, including before eating, and avoid contact with pets' feces. HIV-infected children should avoid contact with reptiles (e.g., snakes, lizards, iguanas, and turtles) and with chicks and ducklings because of the risk for salmonellosis.
Travel. The risk for foodborne and waterborne infections among immunosuppressed, HIV-infected persons is magnified during travel to economically developing countries. HIV-infected children who travel to such countries should avoid foods and beverages that might be contaminated, including raw fruits and vegetables, raw or undercooked seafood or meat, tap water, ice made with tap water, unpasteurized milk and dairy products, and items sold by street vendors. Foods and beverages that are usually safe include steaming hot foods, fruits that are peeled by the traveler, bottled (including carbonated) beverages, and water brought to a rolling boil for 1 minute. Treatment of water with iodine or chlorine might not be as effective as boiling and will not eliminate Cryptosporidia but can be used when boiling is not practical.
Preventing First Episode of Disease
HIV-infected children aged ≤5 years should receive the Hib conjugate vaccine (AII) (Figure 1). Clinicians and other health-care providers should consider use of Hib vaccine among HIV-infected children >5 years old who have not previously received Hib vaccine (AIII) (30,34). For these older children, the American Academy of Pediatrics recommends two doses of any conjugate Hib vaccine, administered at least 1--2 months apart (AIII) (90).
HIV-infected children aged 2--59 months should receive the seven-valent PCV (AII). A four-dose series of PCV is recommended for routine administration to infants at ages 2, 4, 6, and 12--15 months; two or three doses are recommended for previously unvaccinated infants and children aged 7--23 months depending on age at first vaccination (36). Incompletely vaccinated children aged 24--59 months should receive two doses of PCV ≥8 weeks apart. Children who previously received three PCV doses need only one additional dose. Additionally, children aged >2 years should receive the 23-valent pneumococcal polysaccharide vaccine (PPSV) (≥2 months after their last PCV dose), with a single revaccination with PPSV 5 years later (CIII) (36) (see http://www.cdc.gov/vaccines/recs/provisional/downloads/pneumo-Oct-2008-508.pdf for the most updated recommendations). Data are limited regarding efficacy of PCV for children aged ≥5 years and for adults who are at high risk for pneumococcal infection. Administering PCV to older children with high-risk conditions (including HIV-infected children) is not contraindicated. (Figures 1 and 2). One study reported that five-valent PCV is immunogenic among HIV-infected children aged 2--9 years (91). A multicenter study of pneumococcal vaccination in a group of HIV-infected children not administered PCV during infancy demonstrated the safety and immunogenicity of two doses of PCV followed by one dose of PPSV for HAART-treated HIV-infected children aged 2--19 years (including some who had previously received PPSV) (92). In a placebo-controlled trial of a nine-valent PCV among South African children, although vaccine efficacy was somewhat lower among children with than without HIV infection (65% versus 85%, respectively), the incidence of invasive pneumococcal disease was substantially lower among HIV-infected vaccine recipients (63).
HIV-infected children probably are at increased risk for meningococcal disease, although not to the extent they are for invasive S. pneumoniae infection. Although the efficacy of conjugated meningococcal vaccine (MCV) and meningococcal polysaccharide vaccine (MPSV) among HIV-infected patients is unknown, HIV infection is not a contraindication to receiving these vaccines (30). MCV is currently recommended for all children at age 11 or 12 years or at age 13--18 years if not previously vaccinated and for previously unvaccinated college freshmen living in a dormitory (44). A multicenter safety and immunogenicity trial of MCV in HIV-infected 11- to 24-year-olds is under way. In addition, children at high risk for meningococcal disease because of other conditions (e.g., terminal complement deficiencies, anatomic or functional asplenia) should receive MCV if aged 2--10 years (BIII) (41). Although the efficacy of MCV among HIV-infected children is unknown, because patients with HIV probably are at increased risk for meningococcal disease, HIV-infected children who do not fit into the above groups may elect to be vaccinated. Revaccination with MCV is indicated for children who had been vaccinated ≥5 years previously with MPSV (CIII).
Because influenza increases the risk for secondary bacterial respiratory infections (93), following guidelines for annual influenza vaccination for influenza prevention can be expected to reduce the risk for serious bacterial infections in HIV-infected children (BIII) (Figures 1 and 2) (35).
To prevent serious bacterial infections among HIV-infected children who have hypogammaglobulinemia (IgG <400 mg/dL), clinicians should use IVIG (AI). During the pre-HAART era, IVIG was effective in preventing serious bacterial infections in symptomatic HIV-infected children (54), but this effect was most clearly demonstrated only in those not receiving daily trimethoprim--sulfamethoxazole (TMP--SMX) for PCP prophylaxis (55). Thus, IVIG is no longer recommended for primary prevention of serious bacterial infections in HIV-infected children unless hypogammaglobulinemia is present or functional antibody deficiency is demonstrated by either poor specific antibody titers or recurrent bacterial infections (CII).
TMP--SMX administered daily for PCP prophylaxis is effective in reducing the rate of serious bacterial infections (predominantly respiratory) in HIV-infected children who do not have access to HAART (AII) (55,94). Atovaquone combined with azithromycin, which provides prophylaxis for MAC as well as PCP, has been shown in HIV-infected children to be as effective as TMP--SMX in preventing serious bacterial infections and is similarly tolerated (95). However, indiscriminate use of antibiotics (when not indicated for PCP or MAC prophylaxis or other specific reasons) might promote development of drug-resistant organisms. Thus, antibiotic prophylaxis is not recommended solely for primary prevention of serious bacterial infections (DIII).
In developing countries, where endemic deficiency of vitamin A and zinc is common, supplementation with vitamin A and zinc conferred additional protection against bacterial diarrhea and/or pneumonia in HIV-infected children (96,97). However, in the United States, although attention to good nutrition including standard daily multivitamins is an important component of care for HIV-infected children, additional vitamin supplementation above the recommended daily amounts is not recommended (DIII).
Discontinuation of Primary Prophylaxis
A clinical trial, PACTG 1008, demonstrated that discontinuation of MAC and/or PCP antibiotic prophylaxis in HIV-infected children who achieved immune reconstitution (CD4 >15%) while receiving ART did not result in excessive rates of serious bacterial infections (46).
Treatment Recommendations
Treatment of Disease
The principles of treating serious bacterial infections are the same in HIV-infected and HIV-uninfected children. Specimens for microbiologic studies should be collected before initiation of antibiotic treatment. However, in patients with suspected serious bacterial infections, therapy should be administered empirically and promptly without waiting for results of such studies; therapy can be adjusted once culture results become available. The local prevalence of resistance to common infectious agents (i.e., penicillin-resistant S. pneumoniae and MRSA) and the recent use of prophylactic or therapeutic antibiotics should be considered when initiating empiric therapy. When the organism is identified, antibiotic susceptibility testing should be performed, and subsequent therapy based on the results of susceptibility testing (AII).
HIV-infected children whose immune systems are not seriously compromised (CDC Immunologic Category I) (98) and who are not neutropenic can be expected to respond similarly to HIV-uninfected children and should be treated with the usual antimicrobial agents recommended for the most likely bacterial organisms (AIII). For example, for HIV-infected children outside of the neonatal period who have suspected community-acquired bacteremia, bacterial pneumonia, or meningitis, empiric therapy with an extended-spectrum cephalosporin (such as ceftriaxone or cefotaxime) is reasonable until culture results are available (AIII) (85,99). The addition of azithromycin can be considered for hospitalized patients with pneumonia to treat other common community-acquired pneumonia pathogens (M. pneumoniae, C. pneumoniae). If MRSA is suspected or the prevalence of MRSA is high (i.e., >10%) in the community, clindamycin or vancomycin can be added (choice based on local susceptibility patterns) (100,101). Neutropenic children also should be treated with an antipseudomonal drug such as ceftazidime or imipenem, with consideration of adding an aminoglycoside if infection with Pseudomonas spp. is thought likely. Severely immunocompromised HIV-infected children with invasive or recurrent bacterial infections require expanded empiric antimicrobial treatment covering a broad range of resistant organisms similar to that chosen for suspected catheter sepsis pending results of diagnostic evaluations and cultures (AIII).
Initial empiric therapy of HIV-infected children with suspected catheter sepsis should include coverage for both gram-positive and enteric gram-negative organisms, such as ceftazidime, which has anti-Pseudomonas activity, and vancomycin to cover MRSA (AIII). Factors such as response to therapy, clinical status, identification of pathogen, and need for ongoing vascular access, will determine the need and timing of catheter removal.
Monitoring and Adverse Events, Including IRIS
The response to appropriate antibiotic therapy should be similar in HIV-infected and HIV-uninfected children, with a clinical response usually observed within 2--3 days after initiation of appropriate antibiotics; radiologic improvement in patients with pneumonia may lag behind clinical response. Fatal hemolytic reaction to ceftriaxone has been reported in an HIV-infected child with prior ceftriaxone treatment (102). Whereas HIV-infected adults experience high rates of adverse and even treatment-limiting reactions to TMP--SMX, in HIV-infected children, serious adverse reactions to TMP--SMX appear to be much less of a problem (103).
IRIS has not been described in association with treatment of bacterial infections in children.
Management of Treatment Failure
Prevention of Recurrence
Status of vaccination against Hib, pneumococcus, meningococcus, and influenza should be reviewed and updated, according to the recommendations outlined in the section "Preventing First Episode of Disease" (Figures 1 and 2) (AI).
TMP--SMX, administered daily for PCP prophylaxis, and azithromycin or atovaquone-azithromycin, administered for MAC prophylaxis, also may reduce the incidence of drug-sensitive serious bacterial infections in children with recurrent serious bacterial infections. Although administration of antibiotic chemoprophylaxis to HIV-infected children who have frequent recurrences of serious bacterial infections may be considered, caution is required when using antibiotics solely to prevent recurrence of serious bacterial infections because of the potential for development of drug-resistant microorganisms and drug toxicity. In rare situations in which antibiotic prophylaxis is not effective in preventing frequent recurrent serious bacterial infections, IVIG prophylaxis can be considered for secondary prophylaxis (BI).
Discontinuation of Secondary Prophylaxis
As noted earlier, PACTG 1008, demonstrated that discontinuation of MAC and/or PCP antibiotic phylaxis in HIV-infected children who achieved immune reconstitution (CD4 >15%) while receiving antiretroviral therapy did not result in excessive rates of serious bacterial infections (46).
Bartonellosis
Epidemiology
Bartonella is a genus of facultative intracellular bacteria including 21 species, only a few of which have been implicated as human pathogens (104--106). Of these, Bartonella henselae and Bartonella quintana cause a spectrum of diseases specifically in immunocompromised hosts, such as those infected with HIV (107,108). These diseases include bacillary angiomatosis and bacillary peliosis. Immunocompromised persons also are susceptible to Bartonella-associated bacteremia and dissemination to other organ systems. Complications of Bartonella infection are relatively uncommon in the pediatric HIV-infected population (4), although complications in adult immunocompromised hosts also can occur in immunocompromised children with AIDS. Bartonella infections involve an intra-erythrocytic phase that appears to provide a protective niche for the bartonellae leading to persistent and often relapsing infection, particularly in immunocompromised persons (104). A feature of infections with the genus Bartonella is the ability of the bacteria to cause either acute or chronic infection with either vascular proliferative or suppurative manifestations, depending on the immune status of the patient (104).
In the general population, B. henselae typically is associated with cat-scratch disease. Most cases of cat-scratch disease occur in patients aged <20 years (109). A study examining the epidemiology of cat-scratch disease in the United States estimated that 437 pediatric hospitalizations associated with cat-scratch disease occurred among children aged <18 years during 2000, giving a national hospitalization rate of 0.6 per 100,000 children aged <18 years and 0.86 per 100,000 children aged <5 years (110). Data are lacking on the epidemiology of infection with Bartonella spp. in HIV-infected children.
The household cat is a major vector for transmission of B. henselae to humans. Transmission of B. henselae from cat to cat appears to be facilitated by cat fleas, but data do not suggest that B. henselae is efficiently transmitted from cats to humans by fleas (111). More than 90% of patients with cat-scratch disease have a history of recent contact with cats, often kittens (109), and a cat scratch or bite (112) has been implicated as the principal mode of cat-to-human transmission. Compared with adult cats, kittens (<1 year of age) are more likely to have B. henselae bacteremia and to have high levels of bacteremia, and more likely to scratch. Despite the evidence against fleaborne cat-to-human transmission, researchers acknowledge the potential for such transmission and the need for further investigation (111). Elimination of flea infestation is important in preventing transmission because contamination of cat claws or of a scratch wound with infected flea feces is a possible mechanism for infecting humans (111). Infection occurs more often during the autumn and winter (109,112--114).
B. quintana is globally distributed. The vector for B. quintana is the human body louse. Outbreaks of trench fever have been associated with poor sanitation and personal hygiene, which may predispose individuals to the human body louse (106).
Clinical Manifestations
The clinical manifestations of B. henselae infection are largely determined by the host's immune response. Localized disease (e.g., focal suppurative regional lymphadenopathy such as in typical cat-scratch disease) appears most common in patients with an intact immune system; systemic infection appears more commonly in immunocompromised patients, although systemic disease has also been reported among otherwise normal children (115,116). Clinical manifestations of B. henselae and B. quintana specific to HIV-infected and other immunocompromised patients include bacillary angiomatosis and bacillary peliosis.
Bacillary angiomatosis is a rare disorder that occurs almost entirely in severely immunocompromised hosts (117,118). It is a vascular proliferative disease that has been reported most often in HIV-infected adults who have severe immunosuppression with a median CD4 count of <50 cells/mm3 in a majority of case studies of HIV-infected adults (108,119). The disease is characterized by cutaneous and subcutaneous angiomatous papules; the lesions of this disease can be confused with KS. Lesions are often papular and red with smooth or eroded surfaces; they are vascular and bleed if traumatized. Nodules may be observed in the subcutaneous tissue and can erode through the skin. Less frequently, it may involve organs other than the skin.
Bacillary peliosis is characterized by angiomatous masses in visceral organs; it mainly occurs in severely immunocompromised patients with HIV infection. It is a vasoproliferative condition that contains blood-filled cystic spaces. The organ most commonly affected is the liver (i.e., peliosis hepatis), but the disease also can involve bone marrow, lymph nodes, lungs, and CNS (120--122).
Immunocompromised patients infected with B. henselae or B. quintana can also present with persisting or relapsing fever with bacteremia, and these bacteria should be considered in the differential diagnosis of fever of unknown origin in immunocompromised children with late-stage AIDS (123). Dissemination to almost all organ systems has been described, including bone (e.g., osteomyelitis), heart (e.g., subacute endocarditis), and CNS (e.g., encephalopathy, seizures, neuroretinitis, transverse myelitis) (124). Most patients with visceral involvement have nonspecific systemic symptoms, including fever, chills, night sweats, anorexia and weight loss, abdominal pain, nausea, vomiting, and diarrhea.
Diagnosis
Bartonella spp. are small, gram-negative bacilli. In cases of bacillary angiomatosis and bacillary peliosis, diagnosis is usually made through biopsy with a characteristic histologic picture: clusters of organisms can be demonstrated with Warthin-Starry silver stain of affected tissue. The organisms can be isolated with difficulty from blood or tissue culture using enriched agar; they have been isolated more successfully from specimens from patients with bacillary angiomatosis and peliosis than from patients with typical cat-scratch disease (107). B. henselae, similar to other Bartonella spp., is a fastidious, slow-growing organism; in most cases, colonies first appear after 9--40 days; therefore incubation for up to 6 weeks is recommended (124).
Serologic tests such as indirect fluorescent antibody (IFA) test and enzyme immunoassay (EIA) are also available. The IFA is available at many commercial laboratories and state public health laboratories and through CDC (109). Unfortunately, cross-reactivity among Bartonella spp. and other bacteria, such as Chlamydia psittaci (115), is common, and serologic tests do not accurately distinguish among them. Additionally, the sensitivity of the currently available IFA is lower in immunocompromised than immune-competent patients; 25% of HIV-infected Bartonella culture-positive patients never develop anti-Bartonella (121).
The most sensitive method of diagnosis is with PCR testing of clinical specimens; different procedures have been developed that can discriminate among different Bartonella spp. (125,126). PCR assays are available in some commercial and research laboratories.
Prevention Recommendations
Preventing Exposure
Prevention of bartonellosis should focus on reducing exposure to vectors of the disease, i.e., the body louse (for B. quintana) and cats and cat fleas (for B. henselae). Controlling cat flea infestation and avoiding cat scratches are therefore critical strategies for preventing B. henselae infections in HIV-infected persons. To avoid exposure to B. quintana, HIV-infected patients should avoid and treat infestation with body lice (AII).
HIV-infected persons, specifically those with severe immunosuppression, should consider the potential risks of cat ownership; risks of cat ownership for HIV-infected children should be discussed with caretakers. If a decision is made to acquire a cat, cats <1 year of age should be avoided (BII) (109,123). HIV-infected persons should avoid playing roughly with cats and kittens to minimize scratches and bites and should promptly wash sites of contact if they are scratched or bitten (BIII) (109). Also, cats should not be allowed to lick open wounds or cuts (BIII). No evidence indicates any benefit from routine culturing or serologic testing of cats for Bartonella infection or from antibiotic treatment of healthy, serologically positive cats (DII) (109).
Preventing First Episode of Disease
No evidence exists that supports the use of chemoprophylaxis for bartonellosis, such as after a cat scratch (CIII).
Discontinuing Primary Prophylaxis
Not applicable.
Treatment Recommendations
Treatment of Disease
Management of typical cat-scratch disease in immunocompetent patients is mainly supportive because the disease usually is self-limited and resolves spontaneously in 2--4 months. Enlarged, painful lymph nodes may need to be aspirated. Cat-scratch disease typically does not respond to antibiotic therapy; the localized clinical manifestations of the disease are believed to result from an immunologic reaction in the lymph nodes with few viable Bartonella present by the time a biopsy is performed (104,127). In one double-blind, placebo-controlled study in a small number (N=29) of immunocompetent older children and adults with uncomplicated cat-scratch disease, azithromycin resulted in a more rapid decrease in initial lymph node volume by sonography, although clinical outcomes did not differ (128). Thus, antibiotic treatment usually is not recommended for uncomplicated localized disease.
The in vitro and in vivo antibiotic susceptibilities of Bartonella do not correlate well for a number of antibiotics; for example, penicillin demonstrates in vitro activity but has no in vivo efficacy (104,115). Although no systematic clinical trials have been conducted, antibiotic treatment of bacillary angiomatosis and peliosis hepatis is recommended on the basis of reported experience in clinical case series because severe, progressive, and disseminated disease can occur, and without appropriate therapy, systemic spread can occur and involve virtually any organ (104,108). Guidelines for treating Bartonella infections have been published (104).
The drug of choice for treating systemic bartonellosis is erythromycin or doxycycline (AII) (104,121). Clarithromycin or azithromycin treatment has been associated with clinical response, and either of these can be an alternative for Bartonella treatment (BIII) (129).
For patients with severe disease, intravenous (IV) administration may be needed initially (AIII) (130). Therapy should be administered for 3 months for cutaneous bacillary angiomatosis and 4 months for bacillary peliosis, CNS disease, osteomyelitis, or severe infections, as treatment must be of sufficient duration to prevent relapse (AII) (104,123). Combination therapy with the addition of rifampin to either erythromycin or doxycycline is recommended for immunocompromised patients with acute, life-threatening infections (BIII) (104,123). Because doxycycline has better CNS penetration than does erythromycin, the combination of doxycycline and rifampin is preferred for treating CNS Bartonella infection, including retinitis (AIII).
Endocarditis is most commonly caused by B. quintana, followed by B. hensalae, but also has been linked with infection with B. elizabethae, B. vinsonii subspecies Berkhoffii, B. vinsonii subspecies Arupensis, B. kohlerae, and B. alsatica (131). For suspected (but culture-negative) Bartonella endocarditis, 14 days of aminoglycoside treatment (AII) accompanied by ceftriaxone (to adequately treat other potential causes of culture-negative endocarditis) with or without doxycycline for 6 weeks is recommended (BII) (104). For documented culture-positive Bartonella endocarditis, doxycycline for 6 weeks plus gentamicin intravenously for the first 14 days is recommended (BII) (104,109).
Penicillins and first-generation cephalosporins have no in vivo activity and should not be used for treatment of bartonellosis (DII) (132). Quinolones and TMP--SMX have variable in vitro activity and an inconsistent clinical response in case reports (115); as a result, they are not recommended for treatment (DIII).
Monitoring and Adverse Events, Including IRIS
Response to treatment can be dramatic in immunocompromised patients. Cutaneous bacillary angiomatosis skin lesions usually improve and resolve after a month of treatment. Bacillary peliosis responds more slowly than cutaneous angiomatosis, but hepatic lesions should improve after several months of therapy.
Some immunocompromised patients develop a potentially life-threatening Jarisch-Herxheimer--like reaction within hours after institution of antibiotic therapy, and immunocompromised patients with severe respiratory or cardiovascular compromise should be monitored carefully after institution of therapy (104,107).
No cases of Bartonella-associated IRIS have been reported.
Management of Treatment Failure
In immunocompromised patients with relapse, retreatment should be continued for 4--6 months; repeated relapses should be treated indefinitely (AIII) (128). Among patients whose Bartonella infections fail to respond to initial treatment, one or more of the second-line regimens should be considered (AIII).
Prevention of Recurrence
Relapses in bone and skin have been reported and are more common when antibiotics are administered for a shorter time (<3 months), especially in severely immunocompromised patients. For an immunocompromised HIV-infected adult experiencing relapse, long-term suppression of infection with doxycycline or a macrolide is recommended as long as the CD4 cell count is <200 cells/mm3 (AIII). Although no data exist for HIV-infected children, it seems reasonable that similar recommendations should be followed (AIII).
Discontinuing Secondary Prophylaxis
No specific data are available regarding the discontinuation of secondary prophylaxis.
Syphilis
Epidemiology
Treponema pallidum can be transmitted from mother to child at any stage of pregnancy or during delivery. Among women with untreated primary, secondary, early latent, or late latent syphilis at delivery, approximately 30%, 60%, 40%, and 7% of infants, respectively, will be infected. Treatment of the mother for syphilis ≥30 days before delivery is required for effective in utero treatment.
Congenital syphilis has been reported despite adequate maternal treatment. Factors that contribute to treatment failure include maternal stage of syphilis (early stage, meaning, primary, secondary, or early latent syphilis), advancing gestational age at treatment, higher Venereal Disease Research Laboratory (VDRL) titers at treatment and delivery, and short interval from treatment to delivery (<30 days) (133,134). In 2005, the rate of congenital syphilis declined to 8 per 100,000 live-born infants (135), down from 14.3 cases per 100,000 in 2000 and 27.9 cases per 100,000 in 1997. Overall, cases of congenital syphilis have decreased 74% since 1996. The continuing decline in the rate of congenital syphilis probably reflects the substantially reduced rate of primary and secondary syphilis among women during the last decade.
Drug use during pregnancy, particularly cocaine use, has been associated with increased risk for maternal syphilis and congenital infection (136). Similarly, HIV-infected women have a higher prevalence of untreated or inadequately treated syphilis during pregnancy, which places their newborns at higher risk for congenital syphilis (137). Mother-to-child HIV transmission might be higher when syphilis coinfection is present during pregnancy (137--139); transmission does not appear to be higher if the mother's syphilis is effectively treated before pregnancy (137).
Although approximately two thirds of sexually transmitted diseases (STDs) diagnosed annually in the United States occur among persons aged <24 years, such individuals account for less than 25% of early syphilis cases. Nevertheless, the prevalence and incidence of syphilis among HIV-infected youth and of HIV infection among youth with syphilis are appreciable; in a study of 320 HIV-infected and uninfected U.S. adolescents aged 12--19 years, the prevalence of syphilis was 9% among HIV-infected girls and 6% among HIV-infected boys (140). In a meta-analysis of 30 studies, the median HIV seroprevalence among persons infected with syphilis in the United States was 15.7% (27.5% among men and 12.4% among women with syphilis) (141).
Clinical Manifestations
Untreated early syphilis during pregnancy can lead to spontaneous abortion, stillbirth, hydrops fetalis, preterm delivery, and perinatal death in up to 40% of pregnancies (142). Among children with congenital syphilis, two characteristic syndromes of clinical disease exist: early and late congenital syphilis. Early congenital syphilis refers to clinical manifestations appearing within the first 2 years of life. Late congenital syphilis refers to clinical manifestations appearing in children >2 years old.
At birth, infected infants may manifest such signs as hepatosplenomegaly, jaundice, mucocutaneous lesions (e.g., skin rash, nasal discharge, mucous patches, condyloma lata), lymphadenopathy, pseudoparalysis of an extremity, anemia, thrombocytopenia, pneumonia, and skeletal lesions (e.g., osteochondritis, periostitis, or osteitis). In a study of 148 infants born to mothers with untreated or inadequately treated syphilis, 47% had clinical, radiographic, or conventional laboratory findings consistent with congenital syphilis, and 44% had a positive rabbit infectivity test, PCR assay, or IgM immunoblot of serum, blood, or CSF (143). However, as many as 60% of infants with congenital syphilis do not have any clinical signs at birth (144). If untreated, these "asymptomatic" infants can develop clinically apparent disease in the ensuing 3 weeks to 6 months. In addition, fever, nephrotic syndrome, and hypopituitarism may occur.
The manifestations of acquired syphilis in older children and adolescents are similar to those of adults (see Guidelines for the Prevention and Treatment of Opportunistic Infections in HIV-Infected Adults) (16). HIV-infected persons with acquired early syphilis might be at increased risk for neurologic complications and uveitis and have higher rates of treatment failure (145).
Diagnosis
The standard serologic tests for syphilis in adults are based on the measurement of IgG antibody. Because IgG antibody in the infant reflects transplacental passively transferred antibody from the mother, interpretation of reactive serologic tests for syphilis among infants is difficult. Therefore, the diagnosis of neonatal congenital syphilis depends on a combination of results from physical, laboratory, radiographic, and direct microscopic examinations.
All infants born to women with reactive nontreponemal and treponemal test results should be evaluated with a quantitative nontreponemal test (e.g., VDRL slide test, rapid plasma reagin [RPR], or the automated reagin test). Neonatal serum should be tested because of the potential for maternal blood contamination of the umbilical cord blood specimens. Specific treponemal tests, such as the fluorescent treponemal antibody absorption (FTA-ABS) test and T. pallidum particle agglutination (TP-PA) test, are not necessary to evaluate congenital syphilis in the neonate. No commercially available IgM test is recommended for diagnostic use. (Note: Some laboratories use treponemal tests, such as EIA, for initial screening, and nontreponemal tests for confirmation of positive specimens (146). However, such an approach with congenital syphilis has not been published.)
Congenital syphilis can be definitively diagnosed if T. pallidum is detected by using darkfield microscopic examination or direct fluorescent antibody staining of lesions or body fluids such as umbilical cord, placenta, nasal discharge, or skin lesion material from the infant. Failure to detect T. pallidum does not definitively rule out infection because false-negative results are common. Pathologic examination of placenta and umbilical cord with specific fluorescent antitreponemal antibody staining is recommended.
Evaluation of suspected cases of congenital syphilis should include a careful and complete physical examination. Further evaluation depends on maternal treatment history for syphilis, findings on physical examination, and planned infant treatment and may include a complete blood count and differential and platelet count, long bone radiographs, and CSF analysis for VDRL, cell count, and protein. HIV-infected infants might have increased cell counts and protein concentrations even in the absence of neurosyphilis. Other tests should be performed as clinically indicated (e.g., chest radiograph, liver-function tests, cranial ultrasound, ophthalmologic examination, and auditory brainstem response).
A proven case of congenital syphilis requires visualization of spirochetes by darkfield microscopy or fluorescent antibody testing of body fluid(s). Finding that an infant's serum quantitative nontreponemal serologic titer that is fourfold higher than the mother's titer suggests infection but is not a criterion in the case definition. A presumptive case of syphilis is defined as maternal untreated or inadequately treated syphilis at delivery, regardless of findings in the infant, or a reactive treponemal test result and signs in an infant of congenital syphilis on physical examination, laboratory evaluation, long bone radiographs, positive CSF VDRL test, or an abnormal CSF finding without other cause.
For diagnosis of acquired syphilis, a reactive nontreponemal test must be confirmed by a specific treponemal test such as FTA-ABS or TP-PA. Treponemal tests usually will remain positive for life, even with successful treatment. The prozone phenomenon (a weakly reactive or falsely negative) reaction might occur more frequently in HIV-infected persons (147). Treponemal antibody titers do not correlate with disease activity and should not be used to monitor treatment response. CSF should be evaluated among HIV-infected adolescents with acquired syphilis of unknown or <1 year's duration or if they have neurologic or ocular symptoms or signs; many clinicians recommend a CSF examination for all HIV-infected patients with syphilis (16).
Prevention Recommendations
Preventing Exposure
Congenital Syphilis
Effective prevention and detection of congenital syphilis depend on the identification of syphilis in pregnant women and, therefore, on the routine serologic screening of pregnant women during the first prenatal visit. In communities and populations in which the risk for congenital syphilis is high, serologic testing and a sexual history also should be obtained at 28 weeks' gestation and at delivery. Moreover, as part of the management of pregnant women who have syphilis, information about treatment of sex partners should be obtained to assess the risk for reinfection. Routine screening of serum from newborns or umbilical cord blood is not recommended. Serologic testing of the mother's serum is preferred over testing of the infant's serum because the serologic tests performed on infant serum can be nonreactive if the mother's serologic test result is of low titer or the mother was infected late in pregnancy. No HIV-exposed infant should leave the hospital unless the maternal serologic status has been documented at least once during pregnancy and at delivery in communities and populations in which the risk for congenital syphilis is high (148,149).
Acquired Syphilis
Primary prevention of syphilis includes routine discussion of sexual behaviors that may place persons at risk for infection. Providers should discuss risk reduction messages that are client-centered and provide specific actions that can reduce the risk for STD acquisition and HIV transmission (150--152).
Routine serologic screening for syphilis is recommended at least annually for all sexually active HIV-infected persons, with more frequent screening (3--6 months) depending on individual risk behaviors (e.g., multiple partners, sex in conjunction with illicit drug use, methamphetamine use, or partners that participate in such activities) (153). Syphilis in an HIV-infected person indicates high-risk behavior and should prompt intensified counseling messages and consideration of referral for behavioral intervention. Persons undergoing screening or treatment for syphilis also should be evaluated for all common STDs (154).
Discontinuing Primary Prophylaxis
Not applicable.
Treatment Recommendations
Treatment of Disease
Penicillin remains the treatment of choice for syphilis, congenital or acquired, regardless of HIV status (AI).
Congenital Syphilis
Data are insufficient to determine whether infants who have congenital syphilis and whose mothers are coinfected with HIV require different evaluation, therapy, or follow-up for syphilis than that recommended for infants born to mothers without HIV coinfection. Response to standard treatment may differ among HIV-infected mothers. For example, some studies in adults have shown a lag in serologic improvement in appropriately treated patients with HIV infection (155).
Infants should be treated for congenital syphilis if the mother has 1) untreated or inadequately treated syphilis (including treatment with erythromycin or any other nonpenicillin regimen), 2) no documentation of having received treatment, 3) receipt of treatment ≤4 weeks before delivery, 4) treatment with penicillin but no fourfold decrease in nontreponemal antibody titer, or 5) fourfold or greater increase in nontreponemal antibody titer suggesting relapse or reinfection (AII) (154). Infants should be treated regardless of maternal treatment history if they have an abnormal examination consistent with congenital syphilis, positive darkfield or fluorescent antibody test of body fluid(s), or serum quantitative nontreponemal serologic titer that is at least fourfold greater than maternal titer (AII) (154).
Treatment for proven or highly probable congenital syphilis (i.e., infants with findings or symptoms or with titers fourfold greater than mother's titer) is aqueous crystalline penicillin G at 100,000--150,000 units/kg/day, administered as 50,000 units/kg/dose intravenously every 12 hours during the first 7 days of life and every 8 hours thereafter for a total of 10 days (AII). If congenital syphilis is diagnosed after 1 month of life, the dosage of aqueous penicillin G should be increased to 50,000 units/kg/dose intravenously every 4--6 hours for 10 days (AII). An alternative to aqueous penicillin G is procaine penicillin G at 50,000 units/kg/dose intramuscularly (IM) daily in a single dose for 10 days (BII). However, aqueous penicillin G is preferred because of its higher penetration into the CSF. No reports have been published of treatment failures with ampicillin or studies of the effectiveness of ampicillin for treating congenital syphilis.
Asymptomatic infants born to mothers who have had adequate treatment and response to therapy, and with a normal physical examination and CSF findings, and who have a serum quantitative nontreponemal serologic titer that is less than fourfold higher than maternal titer might be treated with a single dose of benzathine penicillin G 50,000 units/kg/dose IM with careful clinical and serologic follow-up (BII). However, certain health-care providers would treat such infants with the standard 10 days of aqueous penicillin because physical examination and laboratory test results cannot definitively exclude congenital syphilis in all cases (BII).
Acquired Syphilis
Acquired syphilis in children is treated with a single dose of benzathine penicillin G 50,000 units/kg IM (up to the adult dose of 2.4 million units) for early-stage disease (e.g., primary, secondary, and early latent disease) (AII). For late latent disease, three doses of benzathine penicillin G 50,000 units/kg (up to the adult dose of 2.4 million units) should be administered IM once weekly for three doses (total 150,000 units/kg, up to the adult total dose of 7.2 million units) (AIII). Alternative therapies (e.g., doxycycline, ceftriaxone, or azithromycin) have not been evaluated among HIV-infected patients and should not be used as first-line therapy (EIII) (154). Neurosyphilis should be treated with aqueous penicillin G 200,000--300,000 units/kg intravenously every 4--6 hours (maximum dosage: 18--24 million units/day) for 10--14 days (AII). See Guidelines for the Prevention and Treatment of Opportunistic Infections in HIV-Infected Adults for dosing recommendations for older HIV-infected adolescents with acquired syphilis (16).
Monitoring and Adverse Events, Including IRIS
All seroreactive infants (or infants whose mothers were seroreactive at delivery) should receive careful follow-up examinations and serologic testing (i.e., a nontreponemal test) every 2--3 months until the test becomes nonreactive or the titer has decreased fourfold (AIII). Nontreponemal antibody titers should decline by age 3 months and should be nonreactive by age 6 months if the infant was not infected (i.e., if the reactive test result was caused by passive transfer of maternal IgG antibody) or was infected but adequately treated. The serologic response after therapy might be slower for infants treated after the neonatal period. Whether children with congenital syphilis who also are HIV-infected take longer to become nonreactive and require retreatment is not known.
Treponemal tests should not be used to evaluate treatment response because the results for an infected child can remain positive despite effective therapy. Passively transferred maternal treponemal antibodies can be present in an infant until age 15 months. A reactive treponemal test after age 18 months is diagnostic of congenital syphilis. If the nontreponemal test is nonreactive at this time, no further evaluation or treatment is necessary. If the nontreponemal test is reactive at age 18 months, the infant should be fully (re)evaluated and treated for congenital syphilis (AIII).
Infants whose initial CSF evaluations are abnormal should undergo a repeat lumbar puncture approximately every 6 months until the results are normal (AII). A reactive CSF VDRL test or abnormal CSF indices that cannot be attributed to other ongoing illness requires retreatment for possible neurosyphilis.
HIV-infected children and adolescents with acquired early syphilis (i.e., primary, secondary, early latent) should have clinical and serologic response monitored at age 3, 6, 9, 12, and 24 months after therapy (AIII); nontreponemal test titers should decline by at least fourfold by 6--12 months after successful therapy, with examination of CSF and retreatment strongly considered in the absence of such decline. For syphilis of longer duration, follow-up is indicated at 6, 12, and 24 months; fourfold decline should be expected by 12--24 months. If initial CSF examination demonstrated a pleocytosis, repeat lumbar puncture should be conducted at 6 months after therapy, and then every 6 months until the cell count is normal (AIII). Follow-up CSF examinations also can be used to evaluate changes in the VDRL-CSF or CSF protein levels after therapy, but changes in these parameters occur more slowly than changes in CSF cell counts. Data from HIV-infected adults with neurosyphilis suggest that CSF abnormalities might persist for extended times, and close clinical follow-up is warranted (145).
Syphilis in an HIV-infected child (congenital or acquired) manifesting as IRIS has not been reported, and only very rare reports of syphilis-associated IRIS in adults (primarily syphilitic ocular inflammatory disease) have been reported (156).
Management of Treatment Failure
After treatment of congenital syphilis, children with increasing or stable nontreponmenal titers at age 6--12 months or children who are seropostive with any titer at 18 months should be evaluated (e.g., including a CSF examination) and considered for retreatment with a 10-day course of parenteral penicillin (AIII).
The management of failures of treatment of acquired syphilis in older children and adolescents is identical to that in adults (16). Retreatment of patients with early-stage syphilis should be considered for those who 1) do not experience at least a fourfold decrease in serum nontreponemal test titers 6--12 months after therapy, 2) have a sustained fourfold increase in serum nontreponemal test titers after an initial reduction posttreatment, or 3) have persistent or recurring clinical signs or symptoms of disease (BIII). If CSF examination does not confirm the diagnosis of neurosyphilis, such patients should receive 2.4 million units IM benzathine penicillin G administered at 1-week intervals for 3 weeks (BIII). Certain specialists have also recommended a course of aqueous penicillin G IV or procaine penicillin IM plus probenicid (as described above for treatment of neurosyphilis) for all patients with treatment failure, although data to support this recommendation are lacking (CIII). If titers fail to respond appropriately after retreatment, the value of repeat CSF evaluation or retreatment has not been established.
Patients with late-latent syphilis should be retreated if they 1) have clinical signs or symptoms of syphilis, 2) have a fourfold increase in serum nontreponemal test titer, or 3) experience an inadequate serologic response (less than fourfold decline in nontreponemal test titer) within 12--24 months after therapy if initial titer was high (>1:32) (BIII). Such patients should have a repeat CSF examination. If the repeat CSF examination is consistent with CNS involvement, retreatment should follow the neurosyphilis recommendations (AIII); those without a CSF profile indicating CNS disease should receive a repeat course of benzathine penicillin, 2.4 million units IM weekly for 3 weeks (BIII), although certain specialists recommend following the neurosyphilis recommendations in this situation as well (CIII).
Retreatment of neurosyphilis should be considered if the CSF white blood cell count has not decreased 6 months after completion of treatment or if the CSF-VDRL remains reactive 2 years after treatment (BIII).
Prevention of Recurrence
No recommendations have been developed for secondary prophylaxis or chronic maintenance therapy for syphilis in HIV-infected children.
Discontinuing Secondary Prophylaxis
Not applicable.
Mycobacterial Infections
Mycobacterium tuberculosis
Epidemiology
In 2006, of the 13,779 cases of TB reported in the United States, 807 (6%) occurred in children younger than 15 years (157). Overall, during 1993--2001, 12.9% of adults with TB were reported to be coinfected with HIV, compared with 1.1% of all children with TB (158). However, the actual rate of HIV coinfection in U.S. children with TB is unknown because of the very low rate of HIV testing in this population.
Numerous studies have documented the increased risk for TB among HIV-infected adults. Domestic and international studies have documented a similar increased risk for TB among HIV-infected children (159--162). Unlike other AIDS-related OIs, CD4 cell count is not a sufficient indicator of increased risk for TB in HIV-infected children. Congenital TB is rare but has been reported among children born to HIV-infected women with TB (163,164).
Children with TB almost always were infected by an adult in their daily environment, and their disease represents the progression of primary infection rather than the reactivation disease commonly observed among adults (165). Identification and treatment of the source patient and evaluation of all exposed members of the household are particularly important because other secondary TB cases and latent infections with M. tuberculosis often are found. All confirmed and suspected TB cases must be reported to state and local health departments, which will assist in contact evaluation.
Disease caused by Mycobacterium bovis recently reemerged among children in New York City, and M. bovis is a frequent cause of TB in children in San Diego County (166,167). Recent cases have been associated with ingestion of unpasteurized fresh cheese from Mexico (166). Most M. bovis cases in humans are attributable to ingestion of unpasteurized milk or its products, and exposure to this pathogen in the United States is unlikely except from privately imported products. However, human-to-human airborne transmission from persons with pulmonary disease has been confirmed, and its relevance might be increased by HIV infection. The distinction between M. tuberculosis and M. bovis is important for determining the source of infection for a child who has TB and for selecting a treatment regimen: almost all M. bovis isolates are resistant to pyrazinamide.
Disease associated with bacille Calmette-Guerin (BCG), an attenuated version of M. bovis, has been reported in HIV-infected children vaccinated at birth with BCG (168). IRIS associated with BCG also has been reported among children initiating HAART (22,168).
Internationally, drug resistance is a growing obstacle to controlling TB, but in the United States, effective public health approaches to prevention and treatment have reduced the rates of drug resistance. In the United States during 1993--2001, M. tuberculosis resistant to any first-line anti-TB drugs was identified in 15.2% of children who had culture-positive M. tuberculosis, with higher rates among foreign-born children (19.2%) than among U.S.-born children (14.1%) (158). Multidrug-resistant TB (MDR TB) is unusual among U.S.-born children and adults with TB. The prevalence of multidrug resistance (e.g., at least isoniazid and rifampin) was lower: 2.8% in foreign-born children and 1.4% in U.S.-born children with TB. However, the fraction of adult TB patients in the United States that is foreign born is increasing, and such persons are a potential source of drug-resistant infection for their U.S.-born children.
Clinical Manifestations
Once infected, children aged ≤4 years and all HIV-infected children are more likely to develop active TB disease. Usually the clinical features of TB among HIV-infected children are similar to those among children without HIV infection, although the disease usually is more severe (169,170) and can be difficult to differentiate from illnesses caused by other OIs. Pulmonary involvement is evident in most cases and can be characterized by localized alveolar consolidation, pneumonitis, and hilar and mediastinal adenopathy. Concomitant atelectasis might result from hilar adenopathy compressing bronchi or from endobronchial granulomas. HIV-infected children with TB are more likely to be symptomatic (with fever and cough) and have atypical findings, such as multilobar infiltrates and diffuse interstitial disease. Rapidly progressive disease, including meningitis or mycobacterial sepsis, can occur without obvious pulmonary findings. Both HIV infection and young age increase the rate of miliary disease and TB meningitis. Older HIV-infected children and adolescents have clinical features more similar to those in HIV-infected adults, with the typical apical lung infiltrates and late cavitation (171). Approximately 25% of HIV-uninfected children with TB include extrapulmonary disease as a sole or concomitant site, and HIV-infected children may have an even higher rate. The most common sites of extrapulmonary disease among children include the lymph nodes, blood (miliary), CNS, bone, pericardium, and peritoneum (169,172--174).
Diagnosis
The cornerstone of diagnostic methods for latent TB infection (LTBI) is the tuberculin skin test (TST), administered by the Mantoux method. Because children with HIV infection are at high risk for TB, annual testing of this population is recommended to diagnose LTBI (AIII). Among persons with HIV infection, ≥5 mm of induration is considered a positive (diagnostic) reaction. However, among immunocompetent children with active TB disease, approximately 10% have a negative TST result, and HIV-infected children with TB are even more likely to have a negative result. Therefore, a negative TST result should never be relied on for excluding the possibility of TB. The use of control skin antigens at time of purified protein derivative testing to assess for cutaneous anergy is of uncertain value and no longer routinely recommended (DII).
Sensitivity to tuberculin is reduced by severe viral infections, such as wild-type measles. As a precaution, skin testing scheduled around the time of live-virus vaccination should be done at the same time as, or delayed until 6 weeks after vaccination to avoid any potentially suppressed sensitivity to the skin test (AIII).
Two-step skin testing is used for detecting boosted sensitivity to tuberculin in health-care workers and others at the time of entry into a serial testing program for occupational TB exposure. The utility and predictive value of two-step testing have not been assessed for children (with or without HIV infection), and its use is not recommended (DIII).
Recently, ex vivo assays that determine IFN-γ release from lymphocytes after stimulation by highly specific synthetic M. tuberculosis antigens have been developed to diagnose infection (175). QuantiFERON(r)-TB Gold and QuantiFERON-TB Gold In-Tube (Cellestis Limited, Valencia, California) and the T-SPOT(r).TB assay (Oxford Immunotec, Marlborough, Massachusetts) are now Food and Drug Administration (FDA)-approved and available in the United States. These tests were more specific than the TST in studies among adults, especially among those who are BCG vaccinated. However, as with the TST, these tests are less sensitive in HIV-infected adults with advanced immune suppression (176). In addition, limited data suggest these tests, particularly QuantiFERON, might have less sensitivity for diagnosing infection in young children (177). Their routine use for finding LTBI or diagnosing TB in HIV-infected children is not recommended because of uncertainty about test sensitivity (DIII) (175).
Patients with a positive test for LTBI should undergo chest radiography and clinical evaluation to rule out active disease. Diagnostic microbiologic methods for TB consist of microscopic visualization of acid-fast bacilli from clinical specimens, nucleic-acid amplification for direct detection in clinical specimens, the isolation in culture of the organism, and drug-susceptibility testing, and genotyping. Although acid-fast stained sputum smears are positive in 50%--70% of adults with pulmonary TB, young children with TB rarely produce sputum voluntarily and typically have a low bacterial load (178). Smear results frequently are negative, even among older children who can expectorate and provide a sample (158). Nevertheless, a positive smear result usually indicates mycobacteria, although it does not differentiate M. tuberculosis from other mycobacterial species. Mycobacterial culture improves sensitivity and permits species identification, drug-susceptibility testing, and genotyping. Confirming M. tuberculosis infection with a culture can have greater significance for HIV-infected children because of the difficulties of the differential diagnosis. Therefore, all samples sent for microscopy should be cultured for mycobacteria. Bronchoscopy will increase the likelihood of obtaining a positive smear and culture. Obtaining early-morning gastric aspirates for acid-fast--bacilli stain and culture is the diagnostic method of choice for children unable to produce sputum. A standardized protocol that includes testing of three samples obtained separately may improve the yield from gastric aspirates to 50% (179). Others have shown the potential utility of induced sputum (180,181) and nasopharyngeal aspirates (182) of obtaining diagnostic specimens from children in the outpatient setting.
Two commercial nucleic acid amplification kits are FDA approved for direct detection of M. tuberculosis in sputum samples with positive smear-microscopy results. One of the methods also is approved for sputa with negative microscopy. A positive result from these methods immediately confirms the diagnosis. However, when these tests are used for other specimens, such as gastric aspirates or CSF, sensitivity and specificity have been disappointing (183--185). These assays provide adjunctive, but not primary, diagnostic evaluation of children with TB because a negative result does not rule out TB as a diagnostic possibility and a positive result, unlike culture, does not allow for drug-susceptibility testing. However, it might be useful in establishing the diagnosis of TB among HIV-infected children who have unexplained pulmonary disease when both culture and TSTs may be falsely negative.
Because of the difficulty in obtaining a specimen for bacteriologic diagnosis of TB among children, evidence for the diagnosis often involves linking the child to an adult with confirmed TB with a positive TST and an abnormal radiograph or physical examination in the child (178). A high index of suspicion is important. Suspicion for and diagnosis of TB in HIV-infected children is further complicated by the frequent presence of preexisting or coincidental fever, pulmonary symptoms, and radiographic abnormalities (e.g., chronic lymphoid interstitial pneumonitis or coincident pulmonary bacterial infection) and the decreased sensitivity of TST in this population. Strenuous efforts should be made to obtain diagnostic specimens (three each of sputum or gastric aspirate specimens or induced sputum) whenever TB is presumptively diagnosed or when it is suspected.
Because many children do not have culture-proven TB, and the diagnosis of drug resistance may be delayed in source cases, MDR TB should be suspected in children with TB in the following situations (90,186--188):
- A child who is a close contact of an MDR TB patient.
- A child who is a contact with a TB patient who died while undergoing treatment when reasons exist to suspect the disease was MDR TB (i.e., the deceased patient was a contact of another person with MDR TB, had poor adherence to treatment, or had received more than two courses of antituberculosis treatment).
- A child with bacteriologically proven TB who is not responding to first-line drugs administered with direct observation.
- A child exposed to a source case that remains smear- or culture-positive after 2 months of directly observed first-line antituberculosis therapy.
- A child born in or exposed to residents of countries or regions with a high prevalence of drug-resistant TB.
Antimycobacterial drug-susceptibility testing should be performed on the initial M. tuberculosis isolate and on subsequent isolates if treatment failure or relapse is suspected; the radiometric culture system has been adapted to perform rapid sensitivity testing. Before obtaining results of susceptibility testing or if an organism has not been isolated from specimens from the child, the antimycobacterial drug susceptibility of the M. tuberculosis isolate from and treatment history of the source case can be used to define the probable drug susceptibility of the child's organism and to design the empiric therapeutic regimen for the child.
Prevention Recommendations
Preventing Exposure
Children most commonly are infected with M. tuberculosis from exposure in their immediate environment, usually the household. HIV-infected children may have family members dually infected with HIV and TB. Homeless children and children exposed to institutional settings (including prolonged hospitalization) may be at increased risk. Risk factors (e.g., homelessness, incarceration, exposure to institutional settings) of close contacts of HIV-infected children also should be considered. BCG vaccine, which is not routinely administered in the United States and should not be administered to HIV-infected infants and children, has potential to cause disseminated disease (EII) (189).
Preventing First Episode of Disease
In the United States, where TB exposure is uncommon and BCG is not routinely administered, HIV-infected infants and children should have a TST (5-TU purified protein derivitive) at 3 months of age, and children should be tested at HIV diagnosis. HIV-infected children should be retested at least once per year (AIII).
HIV-infected infants and children should be treated for LTBI if they have a positive TST (AI) or exposure to a person who has contagious TB (after exclusion of active TB disease in the infant or child and regardless of the child's TST results) (AII). Duration of preventive therapy for children should be 9 months, and the preferred regimen is isoniazid (10--15 mg/kg/day [AII] or 20--30 mg/kg twice weekly) [BII]). Liver function tests should be performed before start of isoniazid (AII) for HIV-infected children. The child should be further monitored if baseline tests are abnormal; the child has chronic liver disease; or medications include other potentially hepatotoxic drugs, such as acetaminophen and some antiretroviral drugs. If isoniazid resistance is known or suspected in the source case, rifampin for 4--6 months is recommended (BII). A 2-month regimen of rifampin and pyrazinamide was never recommended for children and now is not recommended for any age group because of an increased risk for severe and fatal hepatotoxicity (EII). Children exposed to drug-resistant strains should be managed by an experienced clinician, and the regimen should be individualized on the basis of knowledge about the source-case susceptibility pattern and treatment history.
A randomized, double-blind, controlled trial of isoniazid in HIV-infected children in South Africa was halted when isoniazid administered daily or twice weekly (according to the cotrimoxazole schedule) helped reduce overall mortality (hazard ratio: 0.46; 95% confidence interval [CI]: 0.22--0.95; p = 0.015) (190). These findings were found across all ages and CDC HIV disease classification categories and were independent of TST result; however, the study may not have been adequately powered to detect these differences. These results suggest that HIV-infected children in areas of extremely high burden of TB may benefit from isoniazid preventive therapy irrespective of any known exposure to TB, but this approach is not recommended in the United States because of the low prevalence of TB (DII).
Discontinuing Primary Prophylaxis
Not applicable.
Treatment Recommendations
Treatment of Disease
Empiric TB therapy should be started in HIV-infected infants and children in whom the diagnosis is suspected and continued until the diagnosis is definitively ruled out (AII). The use of directly observed therapy (i.e., a trained worker, and not a family member, watches the patient ingest each dose of medication) decreases rates of relapse, treatment failures, and drug resistance and is recommended for treatment of all children and adolescents with TB in the United States (AII). The principles for treating TB in the HIV-infected child are the same as for the HIV-uninfected child. However, treating TB in an HIV-infected child is complicated by antiretroviral drug interactions with the rifamycins and overlapping toxicities caused by antiretroviral drugs and TB medications. Rifampin is a potent inducer of the CYP3A family of enzymes. Rifabutin is a less potent inducer but is a substrate of this enzyme system.
Tables 4 and 5 provide doses and side effects of TB medications. In the absence of concurrent HAART, initial empiric treatment of TB disease usually should consist of a four-drug regimen (isoniazid, rifampin, pyrazinamide, and either ethambutol or streptomycin) (AI). For the first 2 months of treatment, directly observed therapy should be administered daily (intensive phase). Modifications of therapy should be based on susceptibility testing, if possible. The drug-susceptibility pattern from the isolate of the adult source case can guide treatment when an isolate is not available from the child. If the organism is susceptible to isoniazid, rifampin, and pyrazinamide during the 2-month intensive phase of therapy, ethambutol (or streptomycin) can be discontinued and the intensive phase completed using three drugs (AI).
After the 2-month intensive phase, treatment of M. tuberculosis known to be sensitive to isoniazid and rifampin is continued with isoniazid and rifampin as directly observed therapy two to three times weekly (continuation phase) (AI); daily therapy during the continuation phase also is acceptable (AI). Children with severe immunosuppression should receive only daily or thrice-weekly treatment during the continuation phase because TB treatment regimens with once- or twice-weekly dosing have been associated with an increased rate of rifamycin resistance among HIV-infected adults with low CD4 cell counts; thus twice-weekly dosing should be considered only for children without immune suppression (e.g., CDC Immunologic Category I: CD4 >25% or ≥500 cells/mm3 if aged ≥6 years) (CIII) (98). Ethionamide can be used as an alternative to ethambutol in cases of TB meningitis (CIII) because ethionamide has better CNS penetration than does ethambutol.
For HIV-infected children with active pulmonary TB disease, the minimum recommended duration of antituberculous drug treatment is 6 months, but some experts recommend up to 9 months (AIII) (191). For children with extrapulmonary disease involving the bones or joints, CNS, or miliary disease, the minimum recommended duration of treatment is 12 months (AIII) (90,192). These recommendations assume that the organism is susceptible to the medications, adherence to the regimen has been ensured by directly observed therapy, and the child has responded clinically and microbiologically to therapy.
For HIV-infected children diagnosed with TB disease, anti-TB treatment must be started immediately (AIII). However, treatment of TB during HAART is complicated by unfavorable pharmacokinetic interactions and overlapping toxicities and should be managed by a specialist with expertise in treating both conditions (AIII). Issues to consider when treating both conditions include 1) the critical role of rifampin because of its potent bactericidal properties; 2) rifampin's potent induction of the CYP3A enzyme system that precludes treatment with all protease inhibitors (PIs) but may allow treatment with non-nucleoside reverse transcriptase inhibitors (NNRTIs); 3) the CYP3A induction by rifabutin is less potent but dose adjustments of both rifabutin and possibly the PIs still may be needed, although minimal data are available for children; 4) overlapping toxicities; and 5) the challenges of adhering to a medication regimen that may include seven or more drugs.
Given these challenges, some experts have argued that the role of rifamycins in treating TB is so important that deferral of HAART should be considered until completion of TB therapy (CIII). Others recommend that, to improve adherence and better differentiate potential side effects, treatment of TB in an antiretroviral naïve HIV-infected child should be initiated 2--8 weeks before antiretroviral medications are initiated (CIII). Consideration of which option to take must account for clinical factors, such as clinical stage of HIV, immune status of the child, age, ability to adhere to complicated drug regimens, and other comorbid conditions. For severely immunocompromised children (Immunologic Category 3) (98), earlier initiation of HAART (e.g., 2 weeks after start of antimycobacterial therapy) may be advisable (despite risk for IRIS), whereas delayed initiation of HAART might be considered for children with higher CD4 counts (BII).
The choice of antiretroviral regimen in an HIV-infected child being treated for TB disease is complex, and advice should be obtained from an expert in the treatment of these two diseases. Starting antiretroviral therapy with a NNRTI-based rather than a PI-based regimen is preferred because NNRTI regimens have fewer interactions with rifampin-based TB therapy (BII). However, NNRTIs also are metabolized through the CYP3A enzyme system, and efavirenz and nevirapine are both CYP3A4 enzyme inducers. Efavirenz is the preferred NNRTI in HIV-infected children aged >3 years; and nevirapine is the preferred NNRTI for children aged <3 years, as the dosing for efavirenz in younger children has not been defined and no pediatric formulation exists. No data exist for children on the pharmacokinetics of either drug in combination with rifampin to make specific recommendations about potential need for an increase in dose of the NNRTI. If a PI is used, a ritonavir-boosted PI such as lopinavir/ritonavir is required. No pharmacokinetic data are available to address whether additional ritonavir boosting is needed in children receiving rifampin and lopinavir/ritonavir-based regimens.
For children already receiving antiretroviral therapy in whom TB has been diagnosed, the issues are equally complicated, and require similar considerations. Treatment for TB must be started immediately (AIII), and the child's antiretroviral regimen should be reviewed and altered, if needed, to ensure optimal treatment for both TB and HIV and to minimize potential toxicities and drug-drug interactions. These recommendations are limited because of the paucity of data on the optimal dosing of medications to treat TB in children, especially in HIV-infected children. Guidelines and recommendations exist for dose adjustments necessary in adults treated with rifabutin and PIs, but the absence of data preclude extrapolating these to HIV-infected children being treated for TB. Consultation with an expert in pediatric HIV and TB infection is recommended. More data are needed on the pharmacokinetics of anti-TB medications in both HIV-infected and HIV-uninfected children.
For treatment of drug-resistant TB, a minimum of three drugs should be administered, including two or more bactericidal drugs to which the isolate is susceptible (AII). Regimens can include three to six drugs with varying levels of activity. Children infected with MDR TB (e.g., resistance to at least isoniazid and rifampin) should be managed in consultation with an expert in this condition (AIII). If the strain is resistant only to isoniazid, isoniazid should be discontinued and the patient treated with 9--12 months of a rifampin- or rifabutin-containing regimen (e.g., rifampin, pyrazinamide, and ethambutol) (BII). If the strain is resistant only to rifampin, risk for relapse and treatment failure increases. Rifampin should be discontinued, and a 2-month induction phase of isoniazid, pyrazinamide, ethambutol, and streptomycin should be administered, followed by an additional continuation phase of isoniazid, pyrazinamide, and ethambutol to complete a minimum of 12--18 months of therapy, with the exact length of therapy based on clinical and radiologic improvement (BIII). Among older adolescents with rifampin-monoresistant strains, isoniazid, ethambutol, and a fluoroquinolone can be administered, with pyrazinamide added for the first 2 months (BIII); an injectable agent (e.g., aminoglycoside such as streptomycin or amikacin) also can be included in the first 2--3 months for patients with severe disease (BIII). When the strain is resistant to isoniazid and rifampin (i.e., MDR TB), therapeutic regimens must be individualized on the basis of the resistance pattern, treatment history of the patient or the source case, relative activities of the drugs, extent of disease, and any comorbid conditions. The duration of therapy should be at least 12 months---usually longer. In children who are smear- or culture-positive at treatment initiation, therapy usually should continue for 18--24 months after smear and culture conversion. Among children with paucibacillary disease (e.g., smear- and culture-negative), duration of therapy may be shorter but should be ≥12 months (BIII) (90,193).
Extensively drug-resistant TB (XDR TB) has emerged globally as an important new threat, particularly in persons infected with HIV (194). XDR TB is a strain of TB resistant to isoniazid and rifampin (which defined MDR TB) with additional resistance to any fluoroquinolone and at least one of three injectable drugs: capreomycin, kanamycin, and amikacin (195). Of the 49 cases of XDR TB identified in the United States from 1993 to 2006, one (2%) occurred in a child aged <15 years (195). However, this number possibly underestimates the burden in children because many TB cases in children are not culture-positive; thus, a definitive diagnosis of drug resistance (including MDR or XDR) is not possible. Children with suspected or confirmed XDR TB should be managed in consultation with an expert because such cases are associated with rapid disease progression in the prescence of HIV coinfection and a high death rate.
Adjunctive treatment with corticosteroids is indicated for children who have TB meningitis; dexamethasone lowers mortality and long-term neurologic impairment (AII). These drugs might be considered for children with pleural or pericardial effusions, severe miliary disease, and substantial endobronchial disease (BIII). Antituberculous therapy must be administered concomitantly. Most experts use 1--2 mg/kg/day of prednisone or its equivalent for 6--8 weeks.
Monitoring and Adverse Events, Including IRIS
Monthly monitoring of clinical and bacteriologic response to therapy is important (AII). For children with pulmonary TB, chest radiographs should be obtained after 2--3 months of therapy to evaluate response (AIII). Hilar adenopathy might persist for as long as 2--3 years despite successful antituberculous therapy, and a normal radiograph is not a criterion to discontinue therapy. Follow-up radiographs after completion of therapy are not necessary unless clinical symptoms recur.
Common side effects associated with TB medications are listed in Table 5. Isoniazid is available as syrup, but some specialists advise against using it because the syrup is unstable and frequently causes diarrhea (DIII). Gastric upset during the initial weeks of isoniazid treatment occurs frequently and often can be avoided by having some food in the stomach when isoniazid is administered. Hepatotoxicity is the most common serious adverse effect. It includes subclinical hepatic enzyme elevation, which usually resolves spontaneously during continuation of treatment, and clinical hepatitis that usually resolves when the drug is discontinued. It rarely progresses to hepatic failure, but the likelihood of life-threatening liver damage increases when isoniazid is continued despite hepatitis symptoms. Hepatotoxicity is less frequent in children than in adults, but no age group is risk-free. Transient asymptomatic serum transaminase elevations have been noted in 3%--10% and clinical hepatitis in <1% of children receiving isoniazid; <1% of children required treatment discontinuation (192,196). However, the rate of hepatotoxicity might be greater in children who take multiple hepatotoxic medications and in children who have HIV infection. Pyridoxine (150 mg/day) is recommended for all symptomatic HIV-infected children treated with isoniazid (AII). HIV-infected children on anti-TB medications should have liver enzymes obtained at baseline and monthly thereafter (AIII). If symptoms of drug toxicity develop, a physical examination and liver enzyme measurement should be repeated (AIII). Mild elevations in serum transaminases (e.g., two to three times the upper limit of normal) do not require discontinuation of drugs if other findings are normal (AII), but they do require more frequent rechecks---as often as weekly---until they resolve.
The most ominous toxicity associated with ethambutol is optic neuritis, with symptoms of blurry vision, central scotomata, and red-green color blindness, which is usually reversible and rare at doses of 15--25 mg/kg among children with normal renal function (193). Assessments of renal function, ophthalmoscopy, and (if possible) visual acuity and color vision, should be performed before starting ethambutol and monitored regularly during treatment with the agent (AIII). Hypothyroidism has been associated with ethionamide and periodic (e.g., monthly) monitoring of thyroid hormone serum concentrations is recommended with its use (AIII).
Major adverse effects of aminoglycoside drugs are ototoxicity and nephrotoxicity. Periodic audiometry, monitoring of vestibular function (as possible), and blood urea nitrogen and creatinine are recommended (AIII).
Secondary drugs used to treat resistant TB have not been well studied in children. These medications should be used in consultation with a TB specialist (AIII). Coadministration of pyridoxine (150 mg/day) with cycloserine is recommended (AII). Thiacetazone can cause severe and often fatal reactions among HIV-infected children, including severe rash and aplastic anemia, and should not be used (EIII).
IRIS in patients receiving anti-TB therapy during HAART has been reported in HIV-infected adults (197--199). New onset of systemic symptoms, especially high fever; expanding CNS lesions; and worsening adenopathy, pulmonary infiltrates, or pleural effusions have been reported in HIV-infected adults during HAART up to several months after the start of TB therapy. Such cases also have been reported in children (22,192,200) and should be suspected in children with advanced immune suppression who initiate HAART and subsequently develop new symptoms.
IRIS occurs in two common clinical scenarios. First, in patients who have occult TB before initiation of HAART, TB may have been unmasked by immune recovery after antiretroviral drug initiation. This "unmasking IRIS" or incident TB-IRIS usually occurs within the first 3--6 months after initiation of HAART, and the infectious pathogen typically is detectable. Secondly, IRIS can occur as paradoxical exacerbation of TB after initiation of HAART in a patient already receiving anti-TB treatment through a clinical recrudescence of a successfully treated infection or symptomatic relapse despite initial clinical improvement and continued microbiologic treatment success (i.e., "paradoxical IRIS"); treatment failure associated with microbial resistance or poor adherence must be ruled out.
The literature on IRIS in children consists largely of case reports and small series, so whether IRIS occurs more often in children than in adults is not clear. Persons with mild-to-moderate symptoms of IRIS have been treated symptomatically with nonsteroidal anti-inflammatory drugs while continuing anti-TB and HIV therapies. In certain cases, use of systemic corticosteroids steroids for 1--2 weeks results in improvement during continuation of TB/HIV therapies (CIII) (197--199). However, no controlled trials of the use of corticosteroids have been published. Despite the development of IRIS, TB therapy should not be discontinued.
Management of Treatment Failure
Most children with TB respond well to medical therapy. If response is not good, then adherence to therapy, drug absorption, and drug resistance should be assessed. Mycobacterial culture, drug-susceptibility testing, and antimycobacterial drug levels should be performed whenever possible. Drug resistance should be suspected in any child whose smear or culture fails to convert after 2 months of directly observed anti-TB therapy. In the absence of initial bacteriologic confirmation of disease, failure should be suspected in children whose clinical symptoms (including failure to gain weight) fail to respond and who have radiographic evidence of disease progression on therapy. As described above, drug-resistant TB should be managed in consultation with an expert.
Prevention of Recurrence
Risk for recurrence is rare in children with drug-susceptible TB who are treated under direct observation. If TB recurs, the child is at high risk for drug resistance and should be managed accordingly.
Chronic suppressive therapy is unnecessary for a patient who has successfully completed a recommended regimen of treatment for TB (DII). Secondary prophylaxis is not recommended for children who have had a prior episode of TB. However, HIV-infected children who were treated for LTBI or TB and who again contact contagious TB should be treated for presumed latent infection, after diagnostic evalution excludes current disease.
Discontinuing Secondary Prophylaxis
Not applicable.
Mycobacterium avium Complex Disease
Epidemiology
Mycobacterium avium complex (MAC) refers to multiple related species of nontuberculous mycobacteria (e.g., M. avium, M. intracellulare, M. paratuberculosis) that are widely distributed in the environment. Comprehensive guidelines on the diagnosis, prevention, and treatment of nontuberculous mycobacterial diseases were recently published (201). These guidelines highlight the tremendous advances in laboratory methods in mycobacteriology that have expanded the number of known nontuberculous mycobacterial species from 50 in 1997 to 125 in 2006.
MAC was the second most common OI among children with HIV infection in the United States after PCP during the pre-HAART era, but its incidence has greatly decreased from 1.3--1.8 episodes per 100 person-years during the pre-HAART era to 0.14--0.2 episodes per 100 person-years during the HAART era (3,4). MAC is ubiquitous in the environment and presumably is acquired by routine exposures through inhalation, ingestion, or inoculation (202). A recent population-based study in Florida of adults and children associated soil exposure, along with black race and birth outside the United States, with MAC infection (203). Respiratory and GI colonization can act as portals of entry that can lead to disseminated infection (204).
MAC can appear as isolated lymphadenitis among HIV-infected children. Disseminated infection with MAC in pediatric HIV infection rarely occurs during the first year of life; its frequency increases with age and declining CD4 count, and it is a complication of advanced immunologic deterioration among HIV-infected children (202,205,206). Disseminated MAC can occur at higher CD4 cell counts among younger HIV-infected children than among older children or adults, especially among HIV-infected children aged <2 years.
Clinical Manifestations
Respiratory symptoms are uncommon among HIV-infected children who have disseminated MAC, and isolated pulmonary disease is rare. Early symptoms can be minimal and may precede mycobacteremia by several weeks. Symptoms commonly associated with disseminated MAC infection among children include persistent or recurrent fever, weight loss or failure to gain weight, sweats, fatigue, persistent diarrhea, and persistent or recurrent abdominal pain. Lymphadenopathy, hepatomegaly, and splenomegaly can occur. Laboratory abnormalities include anemia, leukopenia, and thrombocytopenia. Although serum chemistries are usually normal, some children may have elevated alkaline phosphatase or lactate dehydrogenase. These signs and symptoms also are relatively common in the absence of disseminated MAC among HIV-infected children with advanced immunosuppression.
Diagnosis
Procedures used to diagnose MAC in children are the same as those used for HIV-infected adults (16). MAC is definitively diagnosed by isolation of the organism from blood or from biopsy specimens from normally sterile sites (e.g., bone marrow, lymph node). Multiple mycobacterial blood cultures over time may be required to yield a positive result. Use of a radiometric broth medium or lysis-centrifugation culture technique can enhance recovery of organisms from blood.
Histology demonstrating macrophage-containing acid-fast bacilli strongly indicates MAC in a patient with typical signs and symptoms, but culture is essential to differentiate nontuberculous mycobacteria from M. tuberculosis, determine which nontuberculous mycobacterium is causing infection, and perform drug-susceptibility testing. Testing of MAC isolates for susceptibility to clarithromycin or azithromycin is recommended (BIII). The BACTEC(tm) method for radiometric susceptibility testing can be used. Susceptibility thresholds for clarithromycin are minimal inhibitory concentrations of ≥32 µg/mL and a minimal inhibitory concentration of ≥256 µg/mL for azithromycin (207).
Prevention Recommendations
Preventing Exposure
MAC is ubiquitous in the environment. Available information does not support specific recommendations regarding exposure avoidance. Person-to-person transmission is not believed to be common.
Preventing First Episode of Disease
The most effective way to prevent disseminated MAC among HIV-infected children is to preserve immune function through use of effective antiretroviral therapy. HIV-infected children who have advanced immunosuppression should be offered prophylaxis against disseminated MAC disease according to the following CD4 count thresholds (AII) (208,209):
- Children aged ≥6 years: <50 cells/mm3
- Children aged 2--5 years: <75 cells/mm3
- Children aged 1--2 years: <500 cells/mm3
- Children aged <1 year: <750 cells/mm3
For the same reasons that clarithromycin and azithromycin are the preferred prophylactic agents for adults, either one should be considered for prophylaxis in children (AII); oral suspensions of both agents are commercially available in the United States. Before prophylaxis is initiated, the child should be evaluated for disseminated MAC disease, which should usually include obtaining a blood culture for MAC (AIII).
Although detecting MAC in stool or respiratory tract may precede disseminated disease, no data support initiating prophylaxis in patients with detectable organisms at these sites in the absence of a blood culture positive for MAC. Therefore, routine screening of respiratory or GI specimens for MAC is not recommended (DIII).
Discontinuing Primary Prophylaxis
On the basis of both randomized controlled trials and observational data, primary prophylaxis for MAC can be safely discontinued in HIV-infected adults who respond to antiretroviral therapy with an increase in CD4 count. In a study of discontinuing OI prophylaxis among HIV-infected children whose CD4 percentages were ≥20% for those aged >6 years and ≥25% for those aged 2--6 years, 63 HIV-infected children discontinued MAC prophylaxis, and no MAC events were observed during ≥2 years of follow up (46). On the basis of both these findings and data from studies in adults, primary prophylaxis can be discontinued in HIV-infected children aged >2 years receiving stable HAART for ≥6 months and experiencing sustained (>3 months) CD4 cell recovery well above the age-specific target for initiation of prophylaxis (e.g., similar to adults, >100 cells/mm3, for children aged ≥6 years; and >200 cells/mm3 for children aged 2--5 years) (BII). No specific recommendations exist for discontinuing MAC prophylaxis in HIV-infected children aged <2 years.
Treatment Recommendations
Treatment of Disease
Disseminated MAC infection should be treated in consultation with a pediatric infectious disease specialist who has expertise in pediatric HIV infection (AIII). Combination therapy with a minimum of two drugs is recommended to prevent or delay the emergence of resistance (AI). Monotherapy with a macrolide results in emergence of high-level drug resistance within weeks.
Improved immunologic status is important for controling disseminated MAC disease; potent antiretroviral therapy should be initiated among children with MAC disease who are antiretroviral naïve. However, the optimal time to start HAART in this situation is unknown; many experts treat MAC with antimycobacterial therapy for 2 weeks before starting HAART to try to minimize IRIS, although whether this makes a difference is unknown (CIII). For children already receiving HAART, HAART should be continued and optimized unless drug interactions preclude the safe concomitant use of antiretroviral and antimycobacterial drugs.
Doses and side effects of MAC medications are included in tables 4 and 5. Initial empiric therapy should include two or more drugs (AI): clarithromycin or azithromycin plus ethambutol. Some experts use clarithromycin as the preferred first agent (AI), reserving azithromycin for patients with substantial intolerance to clarithromycin or when drug interactions with clarithromycin are a concern (AII). Clarithromycin levels can be increased by PIs and decreased by efavirenz, but no data are available to recommend dose adjustments for children. Azithromycin is not metabolized by the cytochrome P450 (CYP450) system; therefore, it can be used without concern for significant drug interactions with PIs and NNRTIs.
Because a study in adults demonstrated a survival benefit with the addition of rifabutin to clarithromycin plus ethambutol, some experts would add rifabutin as a third drug to the clarithromycin/ethambutol regimen (CI); however, drug interactions should be checked carefully, and more intensive toxicity monitoring might be warranted if such drugs are administered concomitantly (AIII). Because rifabutin increases CYP450 activity that leads to increased clearance of other drugs (e.g., PIs and NNRTIs), and toxicity might increase with concomitant administration of drugs, other experts recommend against using this third agent in children (CIII). Guidelines and recommendations exist for dose adjustments necessary in adults treated with rifabutin and PIs, but the absence of data in children precludes extrapolating these to HIV-infected children undergoing treatment for disseminated MAC. No pediatric formulation of rifabutin exists, but the drug can be administered mixed with foods such as applesauce. Safety data are limited from use in 22 HIV-infected children (median age: 9 years) who received rifabutin in combination with two or more other antimycobacterial drugs for treatment of MAC for 1--183 weeks; doses ranged from 4 mg/kg/dose to 18.5 mg/kg/dose, and reported adverse effects were similar to those reported in adults (210).
Monitoring and Adverse Events, Including IRIS
Clinically, most patients improve substantially during the first 4--6 weeks of therapy. A repeat blood culture for MAC should be obtained 4--8 weeks after initiation of antimycobacterial therapy in patients who fail to respond clinically to their initial treatment regimen. Improvement in fever can be expected within 2--4 weeks after initiation of appropriate therapy. However, for those with more extensive disease or advanced immunosuppression, clinical response might be delayed, and elimination of the organism from the blood might require up to 12 weeks of effective therapy.
IRIS in patients receiving MAC therapy during HAART has been reported among HIV-infected adults and children (211--213). New onset of systemic symptoms, especially fever or abdominal pain, leukocytosis, and focal lymphadenitis (cervical, thoracic, or abdominal) associated with preexisting but relatively asymptomatic MAC infection has occurred after start of HAART. Before initiation of HAART among HIV-infected children with low CD4 counts, an assessment for MAC should be considered and treatment provided if MAC is identified. However, recent data indicate that MAC prophylaxis with azithromycin did not prevent IRIS (212). Children with moderate symptoms of IRIS can be treated symptomatically with nonsteroidal anti-inflammatory drugs or, if unresponsive to nonsteroidals, a short course (e.g., 4 weeks) of systemic corticosteroid therapy while continuing to receive HAART (CIII).
Adverse effects from clarithromycin and azithromycin include nausea, vomiting, abdominal pain, abnormal taste, and elevations of liver transaminase levels or hypersensitivity reactions. The major toxicity associated with ethambutol is optic neuritis, with symptoms of blurry vision, central scotomata, and red-green color blindness, which usually is reversible and rare at doses of 15--25 mg/kg among children with normal renal function. Assessments of renal function, ophthalmoscopy, and (if possible) visual acuity and color vision should be performed before starting ethambutol and monitored regularly during treatment with the agent (AIII).
Patients receiving clarithromycin plus rifabutin should be observed for the rifabutin-related development of leukopenia, uveitis, polyarthralgias, and pseudojaundice. Tiny, almost transparent, asymptomatic peripheral and central corneal deposits that do not impair vision have been observed in some HIV-infected children receiving rifabutin as part of a multidrug regimen for MAC (210).
Management of Treatment Failure
Treatment failure is defined as the absence of clinical response and the persistence of mycobacteremia after 8--12 weeks of treatment. Repeat susceptibility testing of MAC isolates is recommended in this situation, and a new multidrug regimen of two or more drugs not previously used and to which the isolate is susceptible should be administered (AIII). Drugs that should be considered for this scenario include rifabutin, amikacin, and a quinolone. In HIV-infected adults, data from treating MAC in HIV-uninfected patients indicate an injectable agent such as amikacin or streptomycin should be considered (CIII) (201). Because dosing of these agents in children can be problematic, drug-resistant disseminated MAC should be treated with input from an expert in this disease (AIII). Optimization of antiretroviral therapy is especially important adjunct to treatment for patients in whom initial MAC therapy has failed.
Prevention of Recurrence
Children with a history of disseminated MAC should be administered lifelong prophylaxis to prevent recurrence (AII).
Discontinuing Secondary Prophylaxis
On the basis of immune reconstitution data in adults and data in children discontinuing primary prophylaxis, some experts recommend discontinuation of secondary prophylaxis in HIV-infected children aged >2 years who have completed ≥12 months of treatment for MAC, who remain asymptomatic for MAC, and who are receiving stable HAART (i.e., HAART not requiring change for viral or immune failure) and have sustained (≥6 months) CD4 cell recovery well above the age-specific target for initiation of primary prophylaxis (e.g., similar to adults, >100 cells/mm3, for children aged >6 years and >200 cells/mm3 for children aged 2--6 years) (CIII). Secondary prophylaxis should be reintroduced if the CD4 count falls below the age-related threshold.
Fungal Infections
Aspergillosis
Epidemiology
Aspergillus spp. are ubiquitous molds that are widespread in soil and grow on plants and decomposing organic materials (214); they are infrequent pathogens in HIV-infected children. The most common species causing aspergillosis is A. fumigatus, followed by A. flavus (215,216). Aspergillosis is rare but often lethal in pediatric AIDS patients; the estimated incidence of invasive aspergillosis in pediatric AIDS patients was 1.5%--3% before widespread use of HAART (217--219), and invasive aspergillosis is believed to be much less prevalent during the post-HAART era. Specific risk factors include low CD4 count, neutropenia, corticosteroid use, concurrent malignancy with chemotherapy, broad-spectrum antibiotic exposure, previous pneumonia and respiratory OIs, and HIV-related phagocytic impairment (217,220--224).
Clinical Manifestations
Invasive pulmonary aspergillosis is the most common presentation among HIV-infected children (223,225,226). Other manifestations include necrotizing tracheobronchitis; pseudomembranous tracheobronchitis; and involvement of CNS, skin, sinuses, middle ear, and mastoid bones (217--221,227). Disseminated aspergillosis has been described rarely (217,228). Invasive pulmonary aspergillosis commonly associated with fever, cough, dyspnea, and pleuritic pain. Acute respiratory distress and wheezing or fungal cast production can occur with necrotizing tracheobronchitis, and stridor can occur with laryngotracheitis (214,217,225). Aspergillus infections of the CNS manifest as single or multiple cerebral abscesses, meningitis, epidural abscess, or subarachnoid hemorrhage (214). Cutaneous aspergillosis typically is associated with contaminated adhesive tapes and arm boards used to secure IV devices (214,217).
Diagnosis
The organism usually is not recoverable from blood (except A. terreus) but is isolated readily from lung, sinus, brain, and skin biopsy specimens (217,222,229). A definitive diagnosis requires relevant clinical signs and symptoms and the histopathologic demonstration of organisms in biopsy specimens obtained from involved sites (e.g., liver or brain). Respiratory tract disease can be presumptively diagnosed in the absence of a tissue biopsy if Aspergillus spp. are recovered from a respiratory sample, compatible signs and symptoms are present, and no alternative diagnosis is identified (90). A serologic assay to detect galactomannan, a molecule in the cell wall of Aspergillus spp., is available commercially but has not been evaluated widely in infants and children. In addition, the assay has higher false-positive results in children (230,231). Therefore, use of galactomannan assays for early detection of aspergillosis is not recommended (DIII).
Radiologic examination plays an important role in diagnosis and follow-up of invasive pulmonary aspergillosis. Chest radiograph demonstrates either a diffuse interstitial pneumonitis or a localized wedge-shaped dense infiltrate representing pulmonary infarction (214,217). Computed tomography (CT) of the chest can be used to identify the halo sign, a macronodule surrounded by a perimeter of ground-glass opacity, which is an early sign of invasive pulmonary aspergillosis (232). Cavitation and air crescent formation shown in chest CT with an aspergilloma appear more frequently in older children and adults than in younger children (233--236).
Prevention Recommendations
Preventing Exposutre
In HIV-infected children who are severely immunosuppressed or neutropenic, considerations for preventing exposure to Aspergillus might include excluding plants and flowers from rooms, avoiding food items such as nuts and spices that often are contaminated, and minimizing application of nonsterile biomedical devices and adhesive tape (214,237--239). Other hospital environmental measures that can help prevent aspergillosis outbreaks include placing suitable barriers between patient-care areas and construction sites; routinely cleaning showerheads, hot water faucets, and air-handling systems; repairing faulty air flow; confining patients to hospital rooms supplied with sterile laminar airflow; and installing high-efficiency particulate air filters (90,240--242).
Preventing First Episode of Disease
The use of chemoprophylaxis for aspergillosis is not recommended in HIV-infected children because of the low incidence of invasive disease and the unknown efficacy of prophylaxis in children, combined with the toxicities of likely agents (DIII) (243--245). Low-dose amphotericin B, itraconazole, or voriconazole prophylaxis has been employed to prevent aspergillosis, with unknown efficacy.
Discontinuing Primary Prophylaxis
Not applicable.
Treatment Recommendations
Treatment of Disease
The recommended treatment for invasive aspergillosis is voriconazole, a second-generation triazole and synthetic derivative of fluconazole (246--249). Data in adults have shown voriconazole to be superior to conventional amphotericin B in treating aspergillosis and to be associated with superior survival (AI) (246). However, data regarding fluconazole for children are limited (BII).
In a compassionate-use program of voriconazole that included 42 immunocompromised children with invasive aspergillosis, voriconazole treatment elicited a complete (43%) or partial (45%) response (250,251). The optimal pediatric dose of voriconazole is not yet known. Children require higher doses (on a mg/kg body weight basis) of voriconazole than do adults to attain similar serum concentrations. The recommended dosage of voriconazole for children is 6--8 mg/kg intravenously or 8 mg/kg orally every 12 hours for two doses, followed by 7 mg/kg intravenously or orally twice daily (AII) (252). For critically ill patients, parenteral administration is recommended (AIII). Therapy is continued for ≥12 weeks, but treatment duration should be individualized for each patient according to clinical response (90). Voriconazole has not been studied in HIV-infected children.
Voriconazole is cleared primarily through three key hepatic microsomal CYP450 enzymes---CYP2C19, CYP2C9, and CYP3A4---with most metabolism mediated through CYP2C19 (253). As a result of a point mutation in the gene encoding CYP2C19, some persons poorly metabolize voriconazole, and others metabolize it extensively; about 3%--5% of whites and blacks are poor metabolizers, whereas 15%--20% of Asians are poor metabolizers (248,253). Drug levels can be as much as fourfold greater in persons who are poor metabolizers than in persons who are homozygous extensive metabolizers. Coadministration of voriconazole with drugs that are potent CYP450 enzyme inducers can significantly reduce voriconazole levels. Voriconazole should be used cautiously with HIV PIs and efavirenz because of potential interactions, and consideration should be given to therapeutic drug monitoring if used concomitantly (CIII).
Amphotericin B, either conventional or a lipid formulation has recommendation level BIII in children (90,254). The standard amphotericin B deoxycholate dosage is 1.0--1.5 mg/kg/day. Lipid formulations of amphotericin B allow administration of higher dosage, deliver higher tissue concentrations of drug to reticuloendothelial organs (e.g., lungs, liver, spleen), have fewer infusion-related side effects and less renal toxicity, but are more expensive; dosing of 5 mg/kg/day is recommended.
Surgical excision of a localized invasive lesion may be warranted, especially in sinus aspergillosis, certain cases of pulmonary aspergillosis with impingement on great vessels or pericardium, hemoptysis from a single focus, and erosion into the pleural space or ribs (BIII).
Monitoring and Adverse Events, Including IRIS
The main side effects of voriconazole are reversible dose-dependent visual disturbances that include a perception of increased brightness and blurred vision that occurs in about one third of patients, elevated hepatic transaminases with higher doses, and occasional skin rash (248); as noted earlier, adverse side effects can result from interactions with PIs. The primary toxicities of amphotericin B include infusion-related fever and chills and nephrotoxicity.
Patients should be monitored for adverse effects related to antifungal agents, especially to amphotericin B. Only one case of aspergillosis-associated IRIS has been described (255).
Management of Treatment Failure
The efficacy of antifungal therapy in invasive aspergillosis is extremely poor. No data are available to guide recommendations for managing treatment failure. For patients in whom treatment failed or who were unable to tolerate voriconazole, amphotericin B should be considered (BIII). Itraconazole for aspergillosis refractory to primary therapy with voriconazole is not recommended because of similar mechanisms of action and possible cross-resistance (DIII).
Caspofungin is approved for adults with invasive aspergillosis who do not improve or do not tolerate standard therapy, and it can be considered for treatment failure in children, although data on this drug are limited in children (CIII). In a pharmacokinetic study in 39 children aged 2--12 years, dosing on a body surface area basis was recommended over a weight-based dosing scheme; 50 mg/m2 body surface area once daily resulted in area-under-the-curve concentrations similar to exposure in adults receiving the standard dosage of 50 mg/day (256). Because of limited bioavailability, caspofungin is available only for IV use.
Combination therapy with caspofungin and voriconazole has been studied in a small number of adults and children with invasive aspergillosis (257--259). For salvage therapy, an additional antifungal agent might be added to current therapy, or combination antifungal drugs from different classes other than the initial regimen can be used (BIII) (257,259--264).
Prevention of Recurrence
For patients with acute leukemia and immunosuppression unrelated to HIV, continuation of antifungal therapy throughout immunosuppression seems to be associated with a more favorable outcome (265). However, no data are available on HIV-infected populations, and hence no recommendations can be made for or against secondary prophylaxis (CIII).
Discontinuing Secondary Prophylaxis
Not applicable.
Candida Infections
Epidemiology
The most common fungal infections among HIV-infected children are caused by Candida spp. Oral thrush and diaper dermatitis occur among 50%--85% of HIV-infected children. Candida albicans is the most common cause of mucosal and esophageal candidiasis. Localized disease caused by Candida is characterized by limited tissue invasion to the skin or mucosa. Examples of localized candidiasis include oropharyngeal and esophageal disease, vulvovaginitis, and diaper dermatitis. Once the organism penetrates the mucosal surface and widespread hematogenous dissemination occurs, invasive candidiasis ensues. This can result in candidemia, meningitis, endocarditis, renal disease, endophthalmitis, and hepatosplenic disease.
Oropharyngeal candidiasis (OPC) continues to be one of the most frequent OIs in HIV-infected children during the HAART era (28% of children), with an incidence rate of 0.93 per 100 child-years (3). The incidence of esophageal or tracheobronchial candidiasis also has decreased from 1.2 per 100 child-years during the pre-HAART era to 0.08 per 100 child-years during the HAART era (2001--2004) (1). Candida esophagitis continues to be seen in children who are not responding to antiretroviral therapy (266,267). Children who develop esophageal candidiasis despite HAART may be less likely to have typical symptoms (e.g., odynophagia and retrosternal pain) or have concomitant OPC (268); during the pre-HAART era, concomitant OPC occurred in 94% of children with candida esophagitis (266). Risk factors for esophageal candidiasis include low CD4 count (<100 cells/mm3), high viral load, and neutropenia (<500 cells/mm3) (1,3,266,267).
Disseminated candidiasis is infrequent among HIV-infected children, but Candida can disseminate from the esophagus particularly when coinfection with herpes simplex virus (HSV) or CMV is present (228,266). Candidemia occurs in up to 12% of HIV-infected children with chronically indwelling central venous catheters for total parental nutrition or IV antibiotics (267,269). Approximately 50% of reported cases of Candida bloodstream infections in HIV-infected children are caused by non-albicans Candida spp., including C. tropicalis, C. pseudotropicalis, C. parapsilosis, C. glabrata, C. kruse, and C. dubliniensis. In one study of Cambodian HIV-infected children on HAART who had candidiasis, seven (75%) of nine isolated C. glabrata were resistant to fluconazole, and three (40%) of seven C. parapsilosis isolated were resistant to more than three azole agents (270). Species-specific epidemiology varies widely by geographic location and hospital. A substantial number of children who develop candidemia have received systemically absorbed oral antifungal azole compounds (e.g., ketoconazole or fluconazole) for control of oral and esophageal candidiasis (267). Early detection and treatment of candidemia can decrease mortality. Overall mortality was 90% in one study in children who had >14 days of fever and symptoms before diagnosis of disseminated infection with Candida spp. (228).
Clinical Manifestations
Clinical manifestations of OPC vary and include pseudomembranous (thrush) and erythematous (atrophic), hyperplastic (hypertrophic), and angular cheilitis. Thrush appears as creamy white curdlike patches with inflamed underlying mucosa that is exposed after removal of the exudate. It can be found on the oropharyngeal mucosa, palate, and tonsils. Erythematous OPC is characterized by flat erythematous lesions on the mucosal surface. Hyperplastic candidiasis comprises raised white plaques on the lower surface of the tongue, palate, and buccal mucosa and cannot be removed. Angular cheilitis occurs as red fissured lesions in the corners of the mouth.
Esophageal candidiasis often presents with odynophagia, dysphagia, or retrosternal pain, and unlike adults, a substantial number of children experience nausea and vomiting. Therefore, children with esophageal candidiasis might present with dehydration and weight loss. Evidence of OPC can be absent among children with esophageal candidiasis, particularly those receiving HAART.
New-onset fever in an HIV-infected child with advanced disease and a central venous catheter is the most common clinical manifestation of candidemia. Renal candidiasis presents with candiduria and ultrasonographically demonstrated renal parenchymal lesions, often without symptoms related to renal disease (267). Candidemia can lead to endogenous endophthalmitis, and ocular examination by an ophthalmologist is warranted in children with bloodstream Candida infection.
Diagnosis
Oral candidiasis can be diagnosed by a potassium hydroxide preparation and culture with microscopic demonstration of budding yeast cells in wet mounts or biopsy specimens. For recurrent or refractory OPC, cultures with in vitro susceptibility testing can be used to guide antifungal treatment (271).
Esophageal candidiasis has a classic cobblestoning appearance on barium swallow. In refractory symptomatic cases, endoscopy should be performed to rule out other causes of refractory esophagitis (e.g., HSV, CMV, MAC, and azole-resistant Candida spp.). Endoscopy might show few small white raised plaques to elevated confluent plaques with hyperemia and extensive ulceration.
Candidemia is best diagnosed with blood cultures using lysis-centrifugation techniques (267) or automated broth-based systems (272). When candidemia is present, depending on clinical suspicions, retinal examination for endophthalmitis, abdominal CT or ultrasound for hepatic or renal involvement, and bone scans for osteomyelitis can be considered.
New diagnostic techniques such as the urine D-arabinitol/
L-arabinitol ratio (273), serum D-arabinitol/creatinine ratio (274), Candida antigen mannan (275), (1,3)-beta-D-gulcan assay (276), and real time PCR (277) are promising diagnostic alternatives under development for early diagnosis of invasive candidiasis in children, but none of these assays have been validated for use in children.
Prevention Recommendations
Prevention of Exposure
Candida organisms are common commensals on mucosal surfaces in healthy persons, and no measures are available to reduce exposure to these fungi.
Preventing First Episode of Disease
Routine primary prophylaxis of candidiasis among HIV-infected infants and children is not indicated, given the low prevalence of serious Candida infections (e.g., esophageal, tracheobronchial, disseminated) during the HAART era and the availability of effective treatment (DIII). Concerns exist about the potential for resistant Candida strains, drug interactions between antifungal and antiretroviral agents, and lack of randomized controlled trials in children (278).
Discontinuing Primary Prophylaxi
Not applicable.
Treatment Recommendations
Treatment of Disease
Oropharyngeal candidiasis
Early, uncomplicated infection can be effectively treated with topical therapy using clotrimazole troches or oral polyenes (such as nystatin or amphotericin B suspension) (BII) (279). Troches should not be used in infants (DIII). Resistance to clotrimazole can develop as a consequence of previous exposure to clotrimazole itself or to other azole drugs; resistance correlates with refractory mucosal candidiasis (280).
Systemic therapy with one of the oral azoles (e.g., fluconazole, ketoconazole, or itraconazole) also is effective for initial treatment of OPC (281,282). Oral fluconazole is more effective than nystatin suspension for initial treatment of OPC in infants; is easier to administer to children than the topical therapies; and is the recommended treatment if systemic therapy is used (AI) (281,283).
Itraconazole solution has comparable efficacy to fluconazole and can be used to treat OPC, although it is less well tolerated than fluconazole (AI) (284). Gastric acid enhances absorption of itraconazole solution; itraconazole solution should be taken without food when possible. Itraconazole capsules and oral solution should not be used interchangeably because, at the same dose, drug exposure is greater with the oral solution than with capsules and absorption of the capsule formulation varies. Ketoconazole absorption also varies, and therefore neither itraconazole capsules nor ketoconazole are recommended for treating OPC if fluconazole or itraconazole solutions are available (DII).
Esophageal disease
Systemic therapy is essential for esophageal disease (AI) and should be initiated empirically among HIV-infected children who have OPC and esophageal symptoms. In most patients, symptoms should resolve within days after the start of effective therapy. Oral or IV fluconazole or oral itraconazole solutions, administered for 14--21 days, are highly effective for treatment of Candida esophagitis (AI) (285). For treatment of OPC, ketoconazole and itraconazole capsules are not recommended because of variable absorption and lower efficacy (DII).
Voriconazole, a newer azole antifungal, or caspofungin, an echinocandin inhibitor of fungal (1,3)-beta-D-glucan synthetase that must be administered intravenously because of limited bioavailability, also are effective in treating esophageal candidiasis in HIV-infected adults (BI) (286--288), but there is little experience with use of these drugs in children. Voriconazole has been used in a limited number of children without HIV infection to treat invasive fungal infections, including esophageal candidiasis or candidemia (251,268). Usually children have been initiated on voriconazole intravenously and then switched to oral administration to complete therapy after stabilization. The optimal pediatric dose of voriconazole is not yet known; children require higher doses (on a mg/kg body weight basis) than do adults to attain similar serum concentrations of voriconazole. The recommended voriconazole dosage for children is 6--8 mg/kg intravenously or 8 mg/kg orally every 12 hours (AII) (252,253). A pharmacokinetic study of caspofungin in immunocompromised children aged 2--17 years without HIV infection demonstrated that 50 mg/m2 body surface area/day (70 mg/day maximum) provides comparable exposure to that obtained in adults receiving a standard 50-mg daily regimen (256). Because of limited experience with both of these drugs in children, data are insufficient to recommend use of voriconazole or caspofungin for esophageal or disseminated candidiasis as first-line therapy (CIII).
Invasive disease
Central venous catheters should be removed when feasible in HIV-infected children with candidemia (AII) (267,271).
Conventional amphotericin B (sodium deoxycholate complex) is the drug of choice for most invasive Candida infections in children, administered once daily intravenously over 1--2 hours (AI). In children who have azotemia or hyperkalemia or are receiving high doses (>1 mg/kg), a longer infusion time of 3--6 hours is recommended (BIII) (289). In children with life-threatening disease, the target daily dose of amphotericin B should be administered from the beginning of therapy (BIII). Duration of therapy in treating candidemia should be determined by the presence of deep tissue foci, clinical response, and presence of neutropenia. Children at high risk for morbidity and mortality should be treated until 2--3 weeks after the last positive blood culture and until signs and symptoms of infection have resolved (AIII) (279). Among children with persistent candidemia despite appropriate therapy, investigation for a deep tissue focus of infection should be conducted (e.g., echocardiogram, renal or abdominal ultrasound). Flucytosine has been used in combination with amphotericin B in some children with severe invasive candidiasis, particularly in those with CNS disease (CIII), but it has a narrow therapeutic index.
Fluconazole has been used as an alternative to amphotericin B to treat invasive disease in children who have not recently received azole therapy (AI) (279). Treatment of invasive candidiasis requires higher doses of fluconazole than are used for mucocutaneous disease. Alternatively, an initial course of amphotericin B therapy can be administered and then carefully followed by completion of a course of fluconazole therapy (BIII). Species identification is necessary when using fluconazole because of intrinsic drug resistance among certain Candida spp. (e.g., C. krusei and C. glabrata). Fluconazole administered to children at 12 mg/kg/day provides exposure similar to standard 400 mg daily dosing in adults. Clearance in older adolescents can be similar to adults, so dosing above 600 mg/day should be employed with caution (290).
Antifungal agents in the echinocandin class, including caspofungin, micafungin, and anidulafungin, have been studied in adults with HIV infection, neutropenic children at risk for fungal infections, and children with documented candidiasis (258,291--296). Because of limited experience in children and no data in HIV-infected children, data are insufficient to recommend these drugs as first-line agents for invasive candidiasis in children (CIII). Data are limited on the use of caspofungin in children with systemic candidiasis. A retrospective report in which caspofungin was administered to 20 children aged ≤16 years who had invasive fungal infections (seven had invasive candidiasis) but not HIV infection, the drug was efficacious and well tolerated (258). In a study of 10 neonates with persistent and progressive candidiasis and unknown HIV status, caspofungin was reported to be an effective alternative therapy (294). Micafungin has been studied in HIV-uninfected, neutropenic children at risk for invasive fungal infections. This drug demonstrates dose-proportional pharmacokinetics and an inverse relation between age and clearance suggesting a need for increased dosage in the young child (295). A study of 19 Japanese HIV-uninfected children aged ≤15 years who had confirmed invasive fungal infections, such as candidiasis, showed that plasma concentration of micafungin dosed at 3 mg/kg body weight was similar to that in adults administered 150 mg per dose (297). Micafungin was administered to premature infants receiving antifungal therapy for a suspected invasive fungal infection. Clearance of the drug in neonates was more than double that in older children and adults (296). Dosages of 10--15 mg/kg/day have been studied in premature neonates, resulting in area-under-the-curve values consistent with an adult dosage of 100--150 mg/day. One pharmacokinetic study of anidulafungin in HIV-uninfected neutropenic children aged 2--17 years showed drug concentrations at 0.75 mg/kg per dose and 1.5 mg/kg per dose were similar to drug concentrations in adults with 50 mg per dose and 100 mg per dose, respectively (298).
Data in adults are limited on use of combination antifungal therapy for invasive candidal infections; combination amphotericin B and fluconazole resulted in more frequent clearance of Candida from the bloodstream but no difference in mortality (299). Data are insufficient to support the routine use of combination therapy in children with invasive candidiasis (DIII) (249).
Monitoring and Adverse Events, Including IRIS
No adverse effects have been reported with the use of oral nystatin for treatment of oral candidiasis, but bitter taste might contribute to poor adherence.
The azole drugs have relatively low rates of toxicity, but because of their ability to inhibit the CYP450-dependent hepatic enzymes (ketoconazole has the strongest inhibitory effect) they can interact substantially with other drugs undergoing hepatic metabolism. These interactions can result in decreased plasma concentration of the azole because of increased metabolism induced by the coadministered drug or development of unexpected toxicity from the coadministered drug because of increased plasma concentrations secondary to azole-induced alterations in hepatic metabolism. The potential for drug interactions, particularly with antiretroviral drugs such as PIs, should be carefully evaluated before initiation of therapy (AIII).
The most frequent adverse effects of the azole drugs are GI, including nausea and vomiting (10%--40% of patients). Skin rash and pruritus might occur with all drugs; rare cases of Stevens-Johnson syndrome and alopecia have been reported with fluconazole therapy. All drugs are associated with asymptomatic increases in transaminases (1%--13% of patients) and, less frequently, hepatitis. Hematologic abnormalities have been reported with itraconazole, including thrombocytopenia and leukopenia. Of the azoles, ketoconazole is associated with the highest frequency of side effects. Its use has been associated with endocrinologic abnormalities related to steroid metabolism, including adrenal insufficiency and gynecomastia, hemolytic anemia, and transaminitis. Dose-related, reversible visual changes (e.g., photophobia and blurry vision) have been reported in approximately 30% of patients receiving voriconazole (300). Cardiac arrhythmias and renal abnormalities including nephritis and acute tubular necrosis also have been reported with voriconazole use.
Amphotericin B deoxycolate undergoes renal excretion as inactive drug. Adverse effects of amphotericin B are primarily nephrotoxicity, defined by substantial azotemia from glomerular damage, and can be accompanied by hypokalemia from tubular damage. Nephrotoxicity is exacerbated by use of concomitant nephrotoxic drugs. Permanent nephrotoxicity is related to cumulative dose. Nephrotoxicity can be ameliorated by hydration before amphotericin B infusion. Infusion-related fevers, chills, nausea, and vomiting occur less frequently in children than in adults. Onset occurs usually within 1--3 hours after the infusion is started, typical duration is <1 hour, and the febrile reactions tend to decrease in frequency over time. Pretreatment with acetaminophen or diphenhydramine might alleviate febrile reactions. Idiosyncratic reactions, such as hypotension, arrhythmias, and allergic reactions, including anaphylaxis, occur less frequently. Hepatic toxicity, thrombophlebitis, anemia, and rarely neurotoxicity (manifested as confusion or delirium, hearing loss, blurred vision, or seizures) also can occur.
In approximately 20% of children, lipid formulations of amphotericin B can cause acute, infusion-related reactions, including chest pain; dyspnea; hypoxia; severe pain in the abdomen, flank, or leg; or flushing and urticaria. Compared with infusion reactions with conventional amphotericin B, most (85%) of the reactions to the lipid formulations occur within the first 5 minutes after infusion and rapidly resolve with temporary interruption of the amphotericin B infusion and administration of IV diphenhydramine. Premedication with diphenhydramine can reduce the incidence of these reactions.
Flucytosine has considerable toxicity: adverse effects on the bone marrow (e.g., anemia, leukopenia, thrombocytopenia), liver, GI tract, kidney, and skin warrant monitoring of drug levels and dose adjustment to keep the level at 40--60 µg/mL. Drug levels should be monitored, especially in patients with renal impairment. High levels can result in bone marrow suppression. The drug should be avoided in children who have severe renal impairment (EIII).
The echinocandins have an excellent safety profile. In a retrospective evaluation of 25 immunocompromised children who received caspofungin, the drug was well tolerated, although three patients had adverse events potentially related to the drug (hypokalemia in all three children, elevated bilirubin in two, and decreased hemoglobin and elevated alanine aminotransferase in one) (256). In this study, children weighing <50 kg received 0.8--1.6 mg/kg body weight daily, and those weighing >50 kg received the adult dosage. In the pharmacokinetic study of 39 children who received caspofungin at 50 mg/m2 body surface area/day, five (13%) patients experienced one or more drug-related clinical adverse events, including one patient each with fever, diarrhea, phlebitis, proteinuria, and transient extremity rash. Two patients reported one or more drug-related laboratory adverse events, including one patient each with hypokalemia and increased serum aspartate transaminase. None of the drug-related adverse events in this study were considered serious or led to discontinuation of caspofungin (256).
IRIS associated with Candida infection has not been described in children. However, evidence suggests that candidiasis occurs with increased frequency in adults during the first 2 months after initiation of HAART, except for candidal eosophagitis (301).
Management of Treatment Failure
Oropharyngeal and esophageal candidiasis
If OPC initially is treated topically, failure or relapse should be treated with oral fluconazole or itraconazole cyclodextrin oral solution (AI) (284,302).
Approximately 50%--60% of patients with fluconazole-refractory OPC and 80% of patients with fluconazole-refractory esophageal candidiasis will respond to itraconazole solution (AII) (303,304). Posaconazole is a second-generation orally bioavailable triazole that has been effective in HIV-infected adults with azole-refractory OPC or esophageal candidiasis (305). However, experience in children is limited, and an appropriate pediatric dosage has not been defined; thus data in children are insufficient to recommend its use in HIV-infected children (CIII) (306,307).
Amphotericin B oral suspension at 1 mL four times daily of a 100-mg/mL suspension sometimes has been effective among patients with OPC who do not respond to itraconazole solution; however, this product is not available in the United States (CIII) (304). Low-dose IV amphotericin B (0.3--0.5 mg/kg/day) has been effective in children with refractory OPC or esophageal candidiasis (BII) (279,304,308,309).
Experience is limited with the use of echinocandins in the treatment of azole-refractory OPC or esophageal candidiasis in children (with or without HIV infection); however, given their excellent safety profile, the echinocandins (306) could be considered for treatment of azole-refractory esophageal candidiasis (CIII).
Invasive disease
Amphotericin B lipid formulations have a role among children who are intolerant of amphotericin B, have disseminated candidal infection that is refractory to conventional amphotericin B, or are at high risk for nephrotoxicity because of preexisting renal disease or use of other nephrotoxic drugs (BII). Although lipid formulations appear to be at least as effective as conventional amphotericin B for treating serious fungal infections (310,311), the drugs are considerably more expensive than conventional amphotericin B. Two lipid formulations are used: amphotericin B lipid complex and liposomal amphotericin B lipid complex. Experience with these preparations among children is limited (254,312,313).
For invasive candidiasis, amphotericin B lipid complex is administered as 5 mg/kg body weight intravenously once daily over 2 hours (254,312,314). Amphotericin B liposome is administered intravenously as 3--5 mg/kg body weight once daily over 1--2 hours. Duration of therapy is based on clinical response; most patients are treated for at least 2--4 weeks.
The role of the echinocandins in invasive candidiasis has not been well studied in HIV-infected children. However, invasive candidiasis associated with neutropenia in patients undergoing bone marrow transplantation has been treated successfully with this class of antifungals. These agents should be considered in the treatment of invasive candidiasis but reserved as alternative, second-line therapy to currently available treatment modalities (CIII).
Prevention of Recurrence
Secondary prophylaxis of recurrent OPC usually is not recommended because 1) the treatment of recurrence is typically effective, 2) the potential exists for the development of resistance and drug interactions, and 3) additional rounds of prophylaxis are costly (DIII). Immune reconstitution with HAART in immunocompromised children should be a priority (AIII). However, if recurrences are severe, data on HIV-infected adults with advanced disease on HAART suggest that suppressive therapy with systemic azoles, either with oral fluconazole (BI) or with itraconazole solution (CI), can be considered (315). Potential azole resistance should be considered when long-term prophylaxis with azoles is considered.
Data on HIV-infected adults suggest that, in children with fluconazole-refractory OPC or esophageal candidiasis who responded to voriconazole or posaconazole therapy or to echinocandins, continuing the effective drug as secondary prophylaxis can be considered because of high relapse rate until HAART produces immune reconstitution (CIII).
Discontinuing Secondary Prophylaxis
In situations where secondary prophylaxis is instituted, no data exist on which to base a recommendation regarding discontinuation. On the basis of experience with HIV-infected adults with other OIs, discontinuation of secondary prophylaxis can be considered when the CD4 count or percentage has risen to CDC Immunologic Category 2 or 1 (CIII) (98).
Coccidioidomycosis
Epidemiology
Coccidioidomycosis is caused by the endemic dimorphic fungus, Coccidioides spp. Two species, Coccidioides posadasii and C. immitis, have been identified using molecular and biogeographic characteristics. C. immitis appears to be confined mainly to California; C. posadasii is more widely distributed through the southwestern United States, northern Mexico, and Central and South America. Most reported infections in these areas represent new infections. Clinical illnesses caused by each are indistinguishable. Infection results from inhalation of spores produced by the fungal form in arid environments with hot summers preceded by rainy seasons (316,317). Infections in regions in which coccidioidomycosis is not endemic usually result from reactivation of a previous infection. Contaminated fomites, such as dusty clothing or agricultural products, also have been implicated as sources of infection (318).
Preexisting impairment of cellular immunity is a major risk factor for severe primary coccidioidomycosis or relapse of past infection. In HIV-infected adults, both localized pneumonia and disseminated infection usually are observed in persons with CD4 counts <250 cells/mm3 (319,320). The threshold for risk in HIV-infected children has not been determined; systemic fungal infection has occurred when CD4 counts were ≤100 cells/mm3 and with CD4 <15%, both indicative of severe immunosuppression (1,15). Although no cases of coccidioidomycosis OI were reported in HIV-infected children from the Perinatal AIDS Collaborative Transmission Study, the study sites are not representative of the areas endemic for coccidioidomycosis (4). Data are limited in children, but in adults, HAART appears to be responsible for the declining incidence of coccidioidomycosis (321).
Clinical Manifestations
Immunocompromised persons and previously healthy blacks, Hispanics, and Filipinos with coccidioidomycosis are at increased risk for dissemination, as are pregnant women who acquire coccidioidal infection during the second or third trimester. Clinical manifestations of coccidioidomycosis in HIV-infected adults are well described. Among these are diffuse pulmonary disease; focal pneumonitis in less immunocompromised patients; extrathoracic dissemination to meninges, lymph nodes, or liver; fever and weight loss syndrome with positive serologies; and the asymptomatic person with positive serologic tests (320). If untreated, a coccidioidal antibody-seropositive, HIV-infected person is at risk for serious disease. Bone and joint involvement is rarely observed in HIV-infected patients (321,322).
Children with primary pulmonary infection may present with fever, malaise, and chest pain. The presence of cough varies, and hemoptysis is rare. Persistent fever may be a sign of dissemination to extrathoracic sites. Children with meningitis may present with headaches, altered sensorium, vomiting, and focal neurologic deficits. Fever is sometimes absent, and meningismus occurs in 50% of patients. Hydrocephalus is common and may occur early. Generalized lymphadenopathy, skin nodules or ulcers, peritonitis, and liver abnormalities also may accompany disseminated disease.
Diagnosis
Because signs and symptoms are nonspecific, coccidioidomycosis should be considered strongly in regions where it is endemic. Coccidioidomycosis also has been reported in regions where it is not endemic, and diagnostic evaluations should be considered in those areas as well.
In patients with meningitis, the CSF shows moderate hypoglycorrhachia, elevated protein concentration, and pleocytosis with a predominance of mononuclear cells. CSF eosinophilia has been reported.
The observation of distinctive spherules containing endospores in histopathologic tissue or clinical specimens is diagnostic. However, stains of CSF in patients with meningitis usually are negative. Pyogranulomatous inflammation with endosporulating spherules is seen readily with hematoxylin and eosin. Spherules can be observed using cytologic staining methods, such as Papanicolau and Gomori methenamine silver nitrate stains. However, cytologic stains are less useful for diagnosing pulmonary coccidioidomycosis than for diagnosing Pneumocystis jirovecii, and a negative cytologic stain on a clinical respiratory specimen does not rule out possible active pulmonary coccidioidomycosis (322). Potassium hydroxide stains are less sensitive and should not be used (DIII) (322).
Growth of Coccidioides spp. is supported by many conventional laboratory media used for fungal isolation at 30°C--37°C (86°F--99°F) with growth occurring within 5 days (322). However, blood cultures are positive in <15% of cases and <50% of CSF from children with meningitis will have a positive culture (322). In contrast, cultures of respiratory specimens are frequently positive in cases of pulmonary coccidioidomycosis in adults.
Although serologic assays that detect coccidioidal-specific antibody are valuable noninvasive aides in diagnosis, negative assays cannot be used to exclude the diagnosis in the immunocompromised host. In the latter instance, detection of coccidioidin or cross-reacting antigens are important diagnostic tests, especially in severe manifestations of disease. Assays for coccidioidal antibody in serum or body fluids such as CSF provide valuable diagnostic and prognostic information. Cross-reactivity may occur with other endemic mycoses. The presence of IgM-specific coccidioidal antibody suggests active or recent infection. The complement fixation (CF) assay detects IgG-specific antibody. CF titers become undetectable in several months if the infection resolves. Standardized CF titers >1:16 directly correlate with the presence and severity of extrapulmonary dissemination. Serologic tests may be falsely negative in severely immunosuppressed HIV-infected children. CF antibody is present in the CSF of 95% of patients with coccidioidal meningitis, but serial testing may be needed to demonstrate this. Titers decline during effective therapy.
Prevention Recommendations
Preventing Exposure
Although HIV-infected persons residing in or visiting regions in which coccidioidomycosis is endemic cannot completely avoid exposure to Coccidioides spp., exposure risk can be reduced by avoiding activities that predispose to inhalation of spores. Such activities include disturbing contaminated soil, excavating archaeologic sites, and being outdoors during dust storms. If such activities are unavoidable, use of respiratory filtration devices should be considered.
Preventing First Episode of Disease
No prospective studies have been published that examine the role of primary prophylaxis to prevent development of active coccidioidomycosis. Although some experts would provide primary prophylaxis with an azole (e.g., fluconazole) to coccidioidal antibody-positive HIV-infected patients living in regions with endemic coccidioidomycosis, others would not (322). Some experts would consider chemoprophylaxis for coccidioidal antibody-positive HIV-infected persons considered at higher risk for active disease, including blacks, persons with unreconstituted cellular immunity with CD4 counts <250 cells/mm3, and persons with a history of thrush (CII) (323). However, given the low incidence of coccidioidomycosis in pediatric HIV-infected patients, possibility of drug interactions, potential antifungal drug resistance, and cost, routine use of antifungal medications for primary prophylaxis of coccidioidial infections in children is not recommended (DIII).
Routine skin testing of HIV-infected patients with coccidioidin (spherulin) does not predict infection and should not be performed (EIII).
Discontinuing Primary Prophylaxis
Not applicable.
Treatment Recommendations
Treatment of Disease
All HIV-infected patients in whom clinically active coccidioidomycosis is diagnosed should be offered antifungal therapy. Treatment protocols for HIV-infected children are based on experience with adults in nonrandomized open-label studies. Physicians who infrequently treat children with coccidioidomycosis should consider consulting with experts.
Because the critical factor in controlling coccidioidomycosis is cellular immune function, effective antiretroviral therapy also is important in treating disease and should be instituted contemporaneously with the initiation of antifungal therapy, if possible.
Diffuse pulmonary or disseminated infection should be treated with amphotericin B deoxycholate at 0.5--1.0 mg/kg/day (AII). Amphotericin B treatment is continued until clinical improvement is observed. The dose and duration of amphotericin B depend on the severity of the symptoms, toxicity, and rapidity of response. Total doses of amphotericin B deoxycholate in adults have ranged from 10 mg/kg to 100 mg/kg. Thereafter, amphotericin B can be discontinued and treatment with fluconazole or itraconazole begun (BIII). Some experts initiate therapy with amphotericin B combined with a triazole, such as fluconazole, in patients with disseminated severe disease and continue the triazole after amphotericin B is stopped (BIII) (322,324). Total duration of therapy should be ≥1 year (322).
No clinical evidence supports greater efficacy of the lipid formulations of amphotericin B than of deoxycholate. However, they are preferred when nephrotoxicity is of concern (BI). A dosage of 5 mg/kg/day is recommended for amphotericin B lipid complex and 3--5 mg/kg/day for liposomal amphotericin B.
For patients with mild disease (such as focal pneumonia), monotherapy with fluconazole or itraconazole is appropriate given their safety, convenient oral dosing, and pharmacodynamic parameters (BII). Thus, fluconazole (5--6 mg/kg/dose twice daily) or itraconazole (5--10 mg/kg/dose twice daily for 3 days followed by 2--5 mg/kg/dose twice daily) are alternatives to amphotericin B for children who have mild, nonmeningitic disease (BIII). In a randomized, double-blind trial in adults, fluconazole and itraconazole were equivalent in treating nonmeningeal coccidioidomycosis. However, itraconazole tended to be superior for skeletal infections (AI) (325).
Treatment of coccidioidal meningitis requires an antifungal agent that achieves high CSF concentrations; thus, IV amphotericin B should not be used (EII). The relative safety and comparatively superior ability of fluconazole to penetrate the blood-brain barrier have made it the azole of choice for coccidioidal meningitis (AII). An effective dose of fluconazole in adults is 400 mg/day (AII), but some experts begin therapy with 800--1000 mg/day (BIII) (324). Children usually receive 5--6 mg/kg/dose twice daily (800 mg/day maximum) (AII) (9). Dosages as high as 12 mg/kg/day have been used (CII) (326). This dosage is required to achieve serum concentrations equivalent to the adult dosage of 400 mg/day (249).
Monitoring and Adverse Events, Including IRIS
In addition to monitoring the patient for clinical improvement, monitoring coccidioidal IgG antibody titers by the complement fixation methodology is useful in assessing response to therapy. Titers should be obtained every 12 weeks (AIII). If therapy is succeeding, titers should decrease progressively, and a rise in titers suggests recurrence of clinical disease. However, if serologic tests initially were negative, titers during effective therapy may increase briefly and then decrease (322). This lag in response during the first 1 or 2 months of therapy should not be construed as treatment failure.
Adverse effects of amphotericin B are primarily nephrotoxicity. Infusion-related fevers, chills, nausea, and vomiting also can occur, although they are less frequent in children than in adults. Lipid formulations of amphotericin B have lower rates of nephrotoxicity. Hepatic toxicity, thrombophlebitis, anemia, and rarely neurotoxicity (manifested as confusion or delirium, hearing loss, blurred vision, or seizures) also can occur (see discussion on monitoring and adverse events in Candida infection).
Triazoles can interact with CYP450-dependent hepatic enzymes, and the potential for drug interactions should be evaluated carefully before initiation of therapy (AIII). Fluconazole and itraconazole appear to be safe in combination with antiretroviral therapy. Voriconazole should be avoided in patients receiving HIV PIs or NNRTIs (320). The most frequent adverse effects of fluconazole are GI, including nausea and vomiting. Skin rash and pruritis might be observed, and rare cases of Stevens-Johnson syndrome have been reported. Asymptomatic increases in transaminases occur in 1%--13% of patients receiving azole drugs. In HIV-infected patients, fluconazole at high doses can cause adrenal insufficiency (327).
Coccidioidomycosis disease in response to IRIS has not been described in children.
Management of Treatment Failure
Clinical information is limited about new therapeutic agents. Posaconazole was effective in six patients with disease refractory to treatment with azoles and amphotericin B (328). Voriconazole was effective in treating coccidioidal meningitis and nonmeningeal disseminated disease in patients who did not respond to fluconazole or were intolerant of amphotericin B (329,330). Caspofungin alone successfully treated disseminated coccidioidomycosis in a renal transplant patient intolerant of fluconazole and in persons in whom conventional therapy failed (331,332). Others have used caspofungin in combination with fluconazole (333).
Adjunctive interferon-gamma was successfully used in a critically ill adult with respiratory failure who did not respond to amphotericin B preparations and fluconazole (334). However, no controlled clinical studies or data exist for children; thus, it is not recommended for use in HIV-infected children (DIII).
Patients with coccidioidal meningitis who do not respond to treatment with the azoles might improve with both systemic amphotericin B and direct instillation of amphotericin B into the intrathecal, ventricular, or intracisternal spaces with or without concomitant azole treatment (CI) (325,326). The basilar inflammation characteristic of coccidioidal meningitis commonly results in obstructive hydrocephalus, necessitating placement of a CSF shunt. Development of hydrocephalus in coccidioidal meningitis does not necessarily indicate treatment failure.
Prevention of Recurrence
Relapse can occur in as many as 33% of patients with disseminated coccidioidomycosis, even in the absence of HIV infection, so lifelong antifungal suppression with either fluconazole or itraconazole is recommended for HIV-infected children who have coccidioidomycosis (AII) (322,324,335--337). In coccidioidal meningitis, response rates to the azoles can be excellent, but cures are infrequent, and relapse after cessation of therapy is common, occurring in as many as 80% of patients (338). Thus, indefinite continuation of fluconazole therapy is recommended in patients who have coccidioidal meningitis (AII).
Discontinuing Secondary Prophylaxis
As with other disseminated fungal infections, continued suppressive therapy with fluconazole or itraconazole is recommended after completion of initial therapy. Patients with diffuse pulmonary disease, disseminated disease, or meningitic disease should remain on lifelong prophylaxis---even if immune reconstitution is achieved with HAART (322)---because of high risk for relapse (AIII). In HIV-infected adults with focal coccidioidal pneumonia who have clinically responded to antifungal therapy and have sustained CD4 count >250 cells/mm3 on HAART, some experts would discontinue secondary prophylaxis after 12 months of therapy with careful monitoring for recurrence with chest radiographs and coccidioidal serology (CIII). However, only a small number of patients have been evaluated, and the safety of discontinuing secondary prophylaxis after immune reconstitution with HAART among children has not been studied extensively. Therefore, for coccidioidomycosis among HIV-infected children, lifelong suppressive therapy is recommended after an acute episode of the disease, regardless of HAART and immune reconstitution (AIII).
Cryptococcosis
Epidemiology
Most cases of cryptococcosis in HIV-infected patients are caused by Cryptococcus neoformans; C. gatti (formerly C. neoformans variety gatti) infection occurs primarily in tropical and subtropical areas. Cryptococcal infections occur much less frequently among HIV-infected children than among adults (339--341). During the pre-HAART era, most cases of cryptococcosis in HIV-infected children (overall incidence, 1%) occurred in those aged 6--12 years and in those with CD4 counts indicating severe immunosuppression (341). Access to HAART has dramatically decreased the overall incidence of cryptococcal infection (342,343), and cryptococcosis in HIV-infected children on HAART remains exceedingly uncommon. Data from various PACTG studies during the pre-HAART and post-HAART eras indicate that the rate of invasive fungal infection, including cryptococcosis, has remained <0.1 per 100 child-years (1,3). In the Perinatal AIDS Collaborative Transmission Study, no cases of cryptococcosis were identified during the HAART era (4).
Clinical Manifestations
Cryptococcosis often presents with subtle and nonspecific findings such as fever and headache. Early diagnosis requires consideration of this infection in a symptomatic patient whose CD4 counts indicate severe immunosuppression. In both HIV-infected adults and children, meningoencephalitis is the most common initial manifestation of cryptococcosis. The disease typically evolves over days to weeks with fever and headache. Less frequent findings include nuchal rigidity, photophobia, and focal neurologic signs, as were seen among 30 HIV-infected children with cryptococcosis reported from the United States (341). In contrast to this indolent presentation, children in Zimbabwe presented with an acute form of neurologic cryptococcosis (69% with nuchal rigidity, 38% with seizure activity, and 23% with focal neurologic signs) (344). CNS mass lesions (cryptococcomas) have not been reported among HIV-infected children.
Disseminated cryptococcosis can be associated with cutaneous lesions, including small, translucent umbilicated papules (indistinguishable from molluscum contagiosum), nodules, ulcers, and infiltrated plaques resembling cellulitis. Diagnosis of pulmonary cryptococcosis without dissemination is unusual among children. Presenting findings include unexplained recurrent fever, cough with scant sputum, intrathoracic lymphadenopathy, and focal or diffuse pulmonary infiltrates. Alternatively the infection may be asymptomatic, with pulmonary nodules revealed on routine chest radiograph (340).
Diagnosis
Detection of cryptococcal antigen in serum, CSF or other body fluids is highly effective for rapid and accurate diagnosis of cryptococcal infection.
Microscopic examination of CSF on India ink-stained wet mounts should be performed to diagnose suspected CNS disease. CSF cell count, glucose, and protein can be virtually normal with CNS cryptococcosis, but the opening pressure usually is elevated. Cryptococcal antigen can be detected in CSF or serum by latex agglutination test (several manufacturers) from >90% of patients with cryptococcal meningitis. However, CSF antigen detection may be negative in culture-positive cryptococcal meningitis; high titers of antigen (prozone effect), low levels of antigen, and nonencapsulated strains contribute to this contradictory result (345,346).
Fungal cultures from CSF, sputum, and blood can identify the organism; the lysis-centrifugation method is the most sensitive for blood specimens. In some cases (e.g., refractory or relapsed disease), susceptibility testing of the C. neoformans isolate can be beneficial. Overall in vitro resistance to antifungal agents remains uncommon (347).
Diffuse pulmonary disease can be diagnosed through bronchoalveolar lavage and direct examination of India ink-stained specimens, culture, and antigen detection. Focal pulmonary and skin lesions may require biopsy with culture and staining.
Prevention Recommendations
Preventing Exposure
No strategies have been proven to prevent exposure. C. neoformans infection is believed to be acquired through inhalation of aerosolized particles from the environment. Serologic studies of immunocompetent children in an urban setting indicate that most children are infected by C. neoformans after the second year of life (348).
Preventing the First Episode of Disease
Because the incidence of cryptococcal disease is so low in HIV-infected children (4,339--341), routine testing of asymptomatic children for serum cryptococcal antigen is not recommended (DIII). Additionally, given the low incidence of cryptococcosis in HIV-infected children, lack of survival benefits in primary prevention studies of adults (349), possibility of drug interaction, potential resistance to antifungal drugs, and cost, routine use of antifungal medications is not recommended for primary prophylaxis of cryptococcal infections in children (DIII).
A review of randomized controlled trials using antifungal interventions for the primary prevention of cryptococcal diseases indicates that fluconazole and itraconazole can reduce cryptococcal disease among adults who have advanced HIV disease and severe immunosuppression (CD4 count <50 cells/mm3) (349). However, neither of these interventions clearly affected mortality.
Discontinuing Primary Prophylaxis
Not applicable.
Treatment Recommendations
Treatment of Disease
Given the low incidence of cryptococcosis in HIV-infected children even during the pre-HAART era, management of this disease in these patients has not been prospectively studied. Treatment recommendations reflect information extrapolated from many well-designed studies involving HIV-infected adults with cryptococcal meningitis.
Unlike immunocompentent hosts, in whom recovery from pulmonary infection without antifungal treatment can occur, immunocompromised hosts with cryptococcosis require treatment because the condition is often fatal in the absence of treatment. Although antifungal treatment is effective, immune reconstitution of the host with the use of antiretroviral medications is crucial to the long-term outcome in terms of avoiding episodes of recurrence and relapse. However, IRIS can be problematic in the short term, posing a diagnostic dilemma when it presents as clinical worsening in a host that was (or is being) treated appropriately for cryptococcal infection. Although data are limited on how best to prevent and manage IRIS, because of the close proximity of OI diagnosis and HAART initiation in patients who developed IRIS (350,351), some clinicians consider delaying HAART in treatment-naïve patients who have treatable OIs until after the acute phase of initial OI therapy has been completed (CIII). Factors other than IRIS, such as tolerability of OI treatments and HAART and overlapping toxicities if both are started concurrently, are also reasons to delay HAART (CIII). No clinical trials have assessed optimal timing of HAART initiation in patients with concurrent cryptococcosis. Overall in vitro resistance to antifungal agents used to treat cryptococcosis remains uncommon (347). Newer azoles (e.g., voriconazole, posaconazole, ravuconazole) are all active in vitro against C. neoformans (347), but published clinical experience in using them for cryptococcosis is limited (352,353).
CNS disease
The most common and well-studied presentation of cryptococcal infection in HIV-infected patients is CNS disease. In light of studies in adults (354--356), combination therapy with amphotericin B and flucytosine for 2 weeks (induction therapy) followed by fluconazole for a minimum of 8 weeks (consolidation therapy) is recommended for children (AI). Cryptococci were cleared from CSF significantly more rapidly from adults with CNS disease who received initial therapy with amphotericin B (0.7 mg/kg/day) and flucytosine (100 mg/kg/day) than it was in those who received amphotericin B alone, amphotericin B plus fluconazole, or triple-antifungal therapy (357,358). In one study of adults, liposomal amphotericin B (AmBisome(r)) dosed at 4 mg/kg/day resulted in significantly earlier CSF culture conversion than did amphotericin B at 0.7 mg/kg/day (359). Although the cost of specific amphotericin B formulations is an important factor in selection of a specific preparation, the indication for use of the drug is also important. For example, the liposomal preparation is preferred for patients with renal insufficiency (AII). Monitoring for and managing raised intracranial pressure is crucial to the optimal management of CNS cryptococcosis (see below).
In patients who cannot tolerate flucytosine, amphotericin B (or its liposomal preparation) alone can be used for initial therapy (BI). Fluconazole plus flucytosine is superior to fluconazole alone (360,361) and provides an alternative to amphotericin B for acute therapy of invasive disease (BII); however, few data are available regarding use of this combination in children, and it should be used only if amphotericin B-based therapy is not tolerated (BIII). Although fluconazole monotherapy was an effective alternative to amphotericin B in adults with AIDS-associated cryptococcal meningitis (362), concerns in this study about differences in early death, delayed CSF sterilization, and drug resistance (363,364) make fluconazole monotherapy less favorable for initial therapy of CNS disease. Because of rapidly developing resistance, flucytosine alone should never be used to treat cryptococcosis (EII).
After a minimum of 2 weeks of induction therapy with evidence of clinical improvement and a negative CSF culture after repeat lumbar puncture, amphotericin B and flucytosine can be discontinued and consolidation therapy initiated with fluconazole (AI). Consolidation therapy is continued for a minimum of 8 weeks (365). Itraconazole is an alternative to fluconazole for the consolidation phase of CNS therapy and for secondary prophylaxis (BI). Fluconazole is preferred because studies comparing the two agents demonstrate higher rates of CSF sterilization during consolidation therapy (355) and less frequent relapse (365) during maintenance therapy in fluconazole recipients.
Pulmonary and extrapulmonary cryptococcosis (CNS disease ruled out)
No controlled clinical studies describe the outcome of non-CNS cryptococcosis in HIV-infected patients. CNS disease should be ruled out in all patients, after which the choice of antifungal medication and length of initial therapy can be decided in light of the clinical severity of illness. Patients with severe pulmonary disease or disseminated cryptococcosis should be treated with amphotericin B with or without the addition of flucytosine, as for CNS disease (AIII). Usually combination therapy should be provided until symptoms resolve. Those with mild-to-moderate pulmonary illness or other localized disease can be managed with fluconazole monotherapy (AIII). Regardless of the antifungal agent selected for initial therapy, secondary prophylaxis with fluconazole or itraconazole should be continued (AIII) until a decision about when to stop prophylaxis can be determined on a case-by-case basis (see note below on discontinuing secondary prophylaxis).
Monitoring and Adverse Events, Including IRIS
Monitoring for raised intracranial pressure
Whenever a lumbar puncture is performed, opening pressure should be measured (AII). Studies in adults clearly show the role of increased intracranial pressure in death associated with CNS cryptococcosis (355,366). Health-care providers should be vigilant about this condition. Its management includes repeated lumbar punctures. In rare cases, CSF shunting may need to be considered for patients who do not tolerate daily lumbar punctures or if standard management does not relieve signs and symptoms of cerebral edema. Corticosteroids and acetazolamide should not be used to reduce intracranial pressure in cryptococcal meningitis (DIII); acetazolamide was associated with severe acidosis, hypokalemia, and other adverse effects in a clinical trial among adults (367).
Monitoring for treatment response
In addition to monitoring clinical response, mycologic response in patients with CNS cryptococcosis typically is assessed by a repeat lumbar puncture and CSF examination at 2 weeks of treatment, with continuation of induction therapy if the culture is positive until negative cultures are obtained.
Monitoring serial serum cryptococcal antigen titers is not useful for following treatment efficacy because changes in serum cryptococcal antigen titers do not correlate well with outcome during treatment for acute meningitis or during suppressive therapy (368,369). Serial measurement of CSF cryptococcal antigen is more useful; in one study, an unchanged or increased titer of antigen in CSF correlated with clinical and microbiologic failure to respond to treatment, and a rise in CSF antigen titer during suppressive therapy was associated with relapse of cryptococcal meningitis (368). However, monitoring of CSF cryptococcal antigen levels requires repeated lumbar punctures and is not routinely recommended for monitoring response (CIII).
Monitoring for adverse events
Adverse effects of amphotericin B (Table 5) are primarily nephrotoxicity; permanent nephrotoxicity is related to cumulative dose. Infusion-related fevers, chills, nausea, and vomiting can occur, but they are less frequent in children than in adults. Close monitoring for drug toxicities is needed especially when amphotericin B is used with flucytosine.
Flucytosine has the potential for marked toxicity, especially affecting the bone marrow (e.g., anemia, leukopenia, and thrombocytopenia), liver, GI tract, kidney, and skin. In patients receiving flucytosine, flucytosine blood levels should be monitored to prevent bone marrow suppression and GI toxicity; peak serum levels, which occur 2 hours after an oral dose, should not exceed 75 µg/mL. Flucytosine should be avoided in children with severe renal impairment (EIII).
Fluconazole and the other azoles have relatively low rates of toxicity, but their potential drug interactions can limit their use. Because of their ability to inhibit the CYP450-dependent hepatic enzymes, the potential for drug interactions, particularly with antiretroviral drugs, should be carefully evaluated before initiation of therapy (AIII).
IRIS
Patients who develop IRIS related to cryptococcosis are more likely than those who do not develop cyrptococcal IRIS to be severely immunocompromised with disseminated infection and to have initiated potent antiretroviral therapy soon after diagnosis of cryptococcal disease (370); in antiretroviral-naïve patients newly diagnosed with cryptococcal meningitis, delay in potent antiretroviral therapy may be prudent until the end of the first 2 weeks of induction therapy (CIII).
IRIS related to cryptococcosis can present within weeks (e.g., meningitis) or months (e.g., lymphadenitis) after start of HAART. Symptoms of meningitis are similar to those described for meningitis presenting as the initial manifestation of cryptococcosis. In one study, about 30% of all HIV-infected adults hospitalized for infection with C. neoformans who received HAART were readmitted with symptoms attributed to an inflammatory response (350). Of the 18 patients with C. neoformans-related IRIS in the cited study, 17 had culture-negative meningitis, and most cases occurred during the first 30 days after initiation of HAART. The most common presentation of late cryptococcal IRIS is lymphadenitis, particularly mediastinal lymphadenitis (371).
The optimal management of cryptococcal IRIS has not been defined. Antifungal therapy should be initiated in patients not already receiving it, and antiretroviral therapy should be continued (AII). Although many cases resolve spontaneously, some experts also have used anti-inflammatory therapy (e.g., short-course corticosteroids) in patients with severely symptomatic IRIS (BIII) (371,372).
Management of Treatment Failure
Treatment failure is defined as clinical deterioration despite appropriate therapy, including management of intracranial pressure; lack of improvement in signs and symptoms after 2 weeks of appropriate therapy; or relapse after an initial clinical response. Differentiating IRIS from treatment failure is important because treatment approaches and outcomes differ. Optimal management of patients with treatment failure is not known. If cultures are positive at treatment failure, evaluation of antifungal susceptibilities might be considered, although fluconazole resistance with C. neoformans is rare in the United States (but more common in some international settings) (363). Patients in whom initial azole-based therapy fails should be switched to amphotericin B-based therapy (363), ideally in combination with flucytosine (BIII); the possibility of drug interactions resulting in subtherapeutic azole levels (e.g., concurrent rifampin use or other drugs metabolized by the liver) should be explored (363). Use of liposomal amphotericin B should be considered, as one study suggests improved efficacy in CSF sterilization with liposomal preparations than with standard amphotericin B (AII) (359). Some data from HIV-infected adults indicate higher dosages (e.g., 400--800 mg/day) of fluconazole in combination with flucytosine also can be considered for salvage therapy (BII) (356,373). Clinical experience with new antifungal agents in managing cryptococcosis is limited. A few patients with cryptococcal infections refractory or intolerant to standard antifungal therapy have been treated with posaconazole or voriconazole with variable success (352,353). Echinocandins do not have clinical activity against cryptococcal infections and should not be used.
Prevention of Recurrence
Patients who have completed initial therapy for cryptococcosis should receive suppressive treatment (i.e., secondary prophylaxis or chronic maintenance therapy) (AI). Fluconazole (AI) is superior and preferable to itraconazole (BI) for preventing relapse of cryptococcal disease (365,374,375). Suppressive therapy typically is continued long term.
Discontinuing Secondary Prophylaxis
Until recently, lifelong secondary prophylaxis typically was recommended. The safety of discontinuing secondary prophylaxis for cryptococcosis after immune reconstitution with HAART has not been studied in children, and decisions in this regard should be made on a case-by-case basis. Adults at apparent low risk for recurrence of cryptococcosis have successfully completed a course of initial therapy, remain asymptomatic with regard to signs and symptoms of cryptococcosis, and have a sustained (≥6 months) increase in their CD4 counts to ≥200 cells/mm3 after HAART (376--378). In light of these observations and inference from data regarding discontinuing secondary prophylaxis for other OIs in adults with advanced HIV infection, discontinuing chronic suppressive therapy for cryptococcosis (after being on it for ≥6 months) can be considered for asymptomatic children aged ≥6 years, on HAART, and with sustained (≥6 months) increase in their CD4 counts to ≥200 cells/mm3 (BII). Suppressive therapy should be reinitiated if the CD4 count decreases to <200 cells/mm3 (AIII).
Histoplasmosis
Epidemiology
Histoplasmosis is caused by inhalation of microconidia produced by the mycelial form of Histoplasma capsulatum, an endemic dimorphic fungus, and cases have been reported from all continents except Antarctica. It is most highly endemic in the Ohio and Mississippi river valleys. Infections in regions in which histoplasmosis is not endemic often result from travel to these regions. Risk factors predisposing to infection are a CD4 count <150 cells/mm3 and exposure to activities that disturb contaminated sites and result in aerosolization of spores. Because yeast forms of the fungus may remain viable within granulomas formed after successful treatment or spontaneous resolution of infection, late relapse can occur if cellular immune function wanes. Infection can occur during pregnancy, and transplacental infection has rarely been reported (379).
During the pre-HAART era, histoplasmosis was reported in 2%--5% of HIV-infected adults in regions with endemic disease; rates of 25% have been reported in some cities (380). In a highly endemic region, histoplasmosis was the AIDS-defining illness in 25% of adults and 8% of children (381). Progressive disseminated histoplasmosis (PDH) occurred in 5% of HIV-infected children in another highly endemic region (M. Kleiman, unpublished data, November 28, 2007). The overall incidence of histoplasmosis in children has not been examined systematically but appeared to be low, even during the pre-HAART era (1).
Few epidemiologic data have been reported on disseminated histoplasmosis in HIV-infected children and adolescents treated with HAART. In several combined PACTG cohorts, the incidence rate of all non-Candida invasive fungal infection was 0.10 infections per 100 child-years (95% CI 0.05--0.20) during the pre-HAART era, and 0.08 infections per 100 child-years (95% CI 0.03--0.17) during the HAART era (1,3). These data were contributed from centers that underrepresented the geographic regions of maximal histoplasmosis prevalence; so the statistical power to detect decreases in incidence rates associated with HAART may have been limited. However, none of the rates of domestic endemic fungal infections (e.g., histoplasmosis, coccidioidomycosis, and blastomycosis) are likely to exceed these estimates in HIV-infected children and adolescents.
Clinical Manifestations
In children without HIV infection, acute pulmonary manifestations are common, but chronic pulmonary infection has not been described. Because of greater airway pliability in children, airway obstruction from mediastinal lymphadenopathy is more common in children (382). Although meningitis is common with progressive disseminated infection in infancy, subacute meningitis and parenchymal lesions characteristic of CNS disease in adults are unusual in children (383). Isolated pulmonary granulomas resulting from past infections are common incidental findings in chest radiographs of asymptomatic persons residing in histoplasmosis-endemic regions.
The most frequent clinical manifestation of histoplasmosis in HIV-infected children with AIDS is PDH; PDH is fatal if untreated. Prolonged fever and failure to thrive are uniform presenting complaints. Few reports have been published of presenting signs and symptoms in children with PDH complicating AIDS (381,384--386). However, most are similar to those seen in PDH in otherwise normal infants and in infections in patients with other primary or acquired immunodeficiencies. These include splenomegaly, cough, respiratory distress, hepatomegaly, "septic" appearance, generalized lymphadenopathy, interstitial pneumonitis, cytopenia, coagulopathy, oropharyngeal/GI ulcerations, and erythematous nodular/ulcerative cutaneous lesions (387--389).
Diagnosis
Culture and histopathologic, serologic, antigen-detection, and molecular diagnostic techniques have been developed to aid in diagnosing histoplasmosis (390,391). Understanding their uses and limitations is essential to interpretating results.
Histoplasmin skin tests are not available and were not useful in diagnosing disseminated disease (388,389). Although isolation of the fungus using culture is diagnostic, it often requires invasive procedures, is insensitive, and may take 10--30 days to grow. Lysis-centrifugation methodology facilitates growth of H. capsulatum, and a DNA probe permits prompt identification of isolates (392). Histopathologic demonstration of typical yeast forms in tissue specimens, bone marrow, or peripheral blood can be performed rapidly and, when positive, is highly suggestive of active infection. However, results are positive in 12%--43% of adults with PDH (390). PCR and DNA probes have been developed to detect H. capsulatum DNA in tissues (393) and body fluids (394) but are neither sufficiently sensitive nor specific (390,391).
Interpretation of serologic testing using CF and immunodiffusion methods is problematic in immunocompromised hosts with PDH. CF titers of ≥1:32 to the yeast and/or mycelial antigens or detection of H and/or M bands with the immunodiffusion test are considered strongly suggestive of active or recent infection. However, only 41% of HIV-infected adults are seropositive, compared with 82% of adults with PDH and no underlying immunocompromise (395). Thus, seronegativity cannot be used to exclude active infection, especially PDH. Although a fourfold increase in CF antibody is diagnostic of active infection, 2--4 weeks is needed to determine this. CF antibody titers of CSF may be useful for diagnosing meningitis. In these instances, the assay should begin with undiluted specimens. Concurrent serum titers should be evaluated to exclude false positivity caused by blood contamination of the CSF (383).
Development and refinement of an EIA that rapidly identifies and quantifies histoplasmal antigen in body fluids fills most of the gaps left by other diagnostic methods. EIA is especially suited for evaluating patients with large fungal burdens, a feature of infection in immunocompromised hosts. EIA can detect antigen in serum, bronchoalveolar lavage, and CSF. The reported sensitivity of antigen detection is 91%--92% in adults with PDH, and 95% in adults with AIDS (390,391); sensitivity in children with underlying immunocompromise and in otherwise normal infants is 100% (388; M Kleiman, unpublished data, November 28, 2007) (396).
EIA has necessitated changes in recommendations for specimen submission and interpretation. In contrast to earlier, semiquantitative assays, the third generation EIA is standardized by extrapolating antigen concentrations from a calibration curve that is linear to a value of 39 ng/mL. However, urine antigen concentrations in serious infections frequently exceed this value. In these instances, serum specimens should be followed because maximum serum concentrations are lower than those of urine and thus more likely to be in a range in which differences can be accurately measured. After resolution of the antigenemia, urine concentrations can be followed to monitor the effectiveness of treatment and, thereafter, to identify relapse. Antigenuria is identified in 90% of patients whose histoplasmosis relapses (382). The histoplasmal antigen assay demonstrates cross-reactions with blastomycosis, paracoccidioidomycosis, and Penicillium marneffei infections (390,391).
Antigen is detectable in 75%--81% of immunocompetent hosts with acute, primary pulmonary infection. This occurs early in infection, reflecting the primary fungemia that is aborted by an effective cellular immune response. Thus, antigenuria in a patient with HIV who retains normal cellular immunity may not presage development of disseminated infection.
Diagnosis of CNS infection is difficult, particularly if the patient has isolated meningitis without disseminated disease (383). Highest sensitivity is achieved by testing CSF for histoplasmal antigen, antibody, and large-volume culture. In adults, CSF culture is positive in 20%--60% of patients, CSF antigen is positive in 40%--70%, and CSF antibody is positive in 70%--90% (390,391); Meningitis frequently accompanies PDH of infancy (387).
Prevention Recommendations
Preventing Exposure
Most infections occur without a recognized history of exposure to a high-risk site or activity. Therefore, complete avoidance of exposure in histoplasmosis-endemic regions is not possible. Sites and conditions commonly implicated in high-risk exposure and point-source outbreaks include soil contaminated with bird or bat droppings, older urban and rural structures, decaying vegetation or trees, and caves. Dry and windy conditions, excavation, demolition, and gardening and agricultural activities predispose to aerosolization of spores. If avoidance of these activities is not feasible, reducing the release of spores by wetting soil, renovation sites, and other likely areas of aerosolization and using protective respiratory devices (397) may reduce the likelihood of infection.
Preventing First Episode of Disease
Prophylaxis with itraconazole is recommended for HIV-infected adults whose CD4 count is <150 cells/mm3 and who reside in areas where histoplasmosis is highly endemic (i.e., incidence is >10 cases per 100 patient-years) and in instances in which risk for occupational exposure is high. Prophylaxis had no effect on survival (382). Given the low incidence of histoplasmosis in pediatric HIV-infected patients, possibility of drug interaction, potential antifungal drug resistance, and cost, routine use of antifungal medications for primary prophylaxis of histoplasma infections in children is not recommended (DIII).
Discontinuing Primary Prophylaxis
Although studies have not been done to support the safety of discontinuing primary prophylaxis, the safety of discontinuing suppressive therapy (secondary prophylaxis) for HIV-infected adults with CD4 counts >150 cells/mm3 has been demonstrated; treatment should be resumed if CD4 count falls below this threshold and the patient continues to reside in an area in which the threshold of >10 cases per 100 patient-years is exceeded (382,398). Prophylaxis is not recommended for HIV-infected children (DIII).
Treatment Recommendations
Treatment of Disease
PDH is fatal without treatment. Therapy with either amphotericin B deoxycholate or itraconazole (399,400) is highly effective. The clinical response to amphotericin B is faster and is preferred for initial treatment of severe infections (AI). Although amphotericin B may be used as monotherapy for an extended time, it is now more commonly used as induction therapy and is followed by long-term treatment with itraconazole. Itraconazole is the azole preferred for treatment of histoplasmosis (AIII). Trials of therapy and the effectiveness of primary and secondary prophylaxis have been evaluated in HIV-infected adults. Recommendations for HIV-infected children are derived from these data and from anecdotal experience in children (382). However, because of important differences in managing PDH in children, consultation with experts should be considered.
The dosage of liposomal amphotericin B for children is 3--5 mg/kg/day. Other less costly or better tolerated lipid formulations may be substituted for the liposomal product. Amphotericin B deoxycholate, at 1 mg/kg/day, is better tolerated by children than it is by adults, is effective, and may be used when cost of the lipid preparations is a consideration.
Itraconazole is usually well tolerated in children. Itraconazole has a long half-life and does not reach steady-state levels for 2 weeks. The interval needed to achieve desired serum concentrations can be shortened if the recommended dose is administered three times daily for the initial 3 days of therapy (i.e., loading dose); the recommended dose administered twice daily should be started thereafter. Itraconazole solution is preferred to the capsule formulation because it is better absorbed and serum concentrations are 30% higher than those achieved with the capsules. Because absorption of itraconazole varies considerably from patient to patient, serum concentrations should be measured to ensure effective levels of drug, monitor changes in dosage, and assess compliance (BIII). The minimal inhibitory concentration of H. capsulatum is 0.01 µg/mL, and although minimally effective serum concentrations have not been determined, a serum concentration of 1.0 µg/mL is recommended; dosage should be reduced if concentrations exceed 10 µg/mL (382).
Fluconazole is an alternative for patients with mild histoplasmosis who are intolerant of itraconazole or in whom desired serum levels of itraconazole cannot be attained. However, fluconazole is less effective than itraconazole and has been associated with development of drug resistance (401). Ketoconazole is used infrequently because of its adverse reactions; it is effective in mild infections, excluding disseminated infection, and may be considered because it is much less costly than the other azoles.
Acute primary pulmonary histoplasmosis
Acute primary pulmonary histoplasmosis can present with a wide spectrum of symptoms ranging from dyspnea with high fever and diffuse pulmonary infiltrates to only mild respiratory symptoms, variable fever, and a chest radiograph showing mediastinal adenopathy with or without focal pulmonary infiltrate. For severe or moderately severe symptoms, amphotericin B should be administered for 1--2 weeks; amphotericin B deoxycholate is preferred because it is well tolerated in children (AIII) (382). After clinical improvement, patients with intact immunity should receive itraconazole, beginning with a loading dose (see above) for the first 3 days, followed by the recommended doses administered twice daily for at least 12 weeks (AIII); adults with CD4 counts of <150 cells/mm3, and by extrapolation, HIV-infected children with severe immunosuppression (e.g., CD4 <15% or <150 cells/mm3 in children aged ≤6 years), should receive itraconazole for 12 months (AIII). Urine antigen usually is elevated in these situations and should be monitored to gauge clinical response and, after treatment, identify relapse (AIII).
HIV-infected children, particularly those with functional cellular immunity, occasionally will present with fever associated with mild primary pulmonary infection and histoplasma antigen in the urine. Although an effective cellular immune response may limit such illnesses, treatment with itraconazole for 12 weeks while following histoplasmal urine antigen concentrations may be prudent to ensure concentrations decrease (BIII).
Moderately severe to severe PDH
Data from HIV-infected adults suggest that HIV-infected children with moderately severe to severe disseminated histoplasmosis should be treated with an IV amphotericin B formulation for ≥2 weeks or until they clinically improve, followed by itraconazole for 12 months (AI). HIV-infected adults with moderately severe to severe PDH have a higher response rate to treatment with liposomal amphotericin B than with the deoxycholate formulation (88% vs 64%) and a lower death rate (2% vs 13%) (AI) (382). After a favorable clinical response, amphotericin B should be discontinued and followed by "step-down" therapy with itraconazole for 12 months (AII). A loading dose (see above) of itraconazole should be used for the initial 3 days. If itraconazole is not well tolerated, a 4- to 6-week course of amphotericin B should be used and histoplasma urine antigen levels followed (AIII).
Although therapeutic trials of amphotericin B deoxycholate used to treat PDH in HIV-infected children have not been performed, this formulation is effective for treating severe PDH in infants (387,402), including those with CNS infection (387), and in children with other primary or acquired immunodeficiency states. Amphotericin B deoxycholate is better tolerated by children than by adults, and it is less costly than other formulations. It may be used if cost or availability of lipid formulations precludes their use (AIII).
Mild to moderate PDH
Mild to moderate PDH in adults without signs of CNS infection responds favorably in 80%--100% of patients treated with itraconazole monotherapy for 12 months (AII) (382,399). This regimen also is recommended for HIV-infected children with mild to moderate PDH (AII). A loading dose of itraconazole (see above) should be administered at the onset of treatment and serum concentrations monitored.
CNS infection
CNS infection that accompanies PDH is expected to respond to the regimen recommended for moderately severe to severe PDH. Isolated CNS infection is unusual in children. In adults, frequent failure and relapse are common, and aggressive therapy is recommended. Liposomal amphotericin B is preferred for CNS disease in children and adults because it achieves higher concentrations in the brain (AII); penetration into the CSF is poor with all formulations. The deoxycholate formulation is an alternative. Amphotericin should be administered for 4--6 weeks. Thereafter, the child should receive a loading dose of itraconazole and continuation of itraconazole for 12 months and until CSF abnormalities, including histoplasmal antigen, have resolved (AII). Itraconazole levels should be followed and dose adjusted to ensure optimal serum concentrations (AIII).
Asymptomatic Histoplasma granuloma
In asymptomatic HIV-infected children who have intact cellular immunity and have resided in an area with endemic histoplasmosis, the presence of a typical granuloma in a chest radiograph should prompt evaluation of both histoplasmal urine antigen and CF and immunodiffusion antibody. If any of these tests are positive, treatment with itraconazole for 12 weeks is prudent (BIII). If these tests are negative, therapy need not be used, and clinical follow-up is recommended. In either instance, histoplasmal urine antigen testing should be considered if unexplained fever or other systemic symptoms occur.
Monitoring and Adverse Events, Including IRIS
In manifestations of histoplasmosis in which antigenuria is demonstrated, antigen levels should be monitored during therapy and for a year thereafter to identify relapse (AIII) (403). After a recommended course of therapy and, in the absence of symptoms, low-level, stable antigenuria may not constitute a basis for prolonging the recommended course of therapy. Serum levels of itraconazole should be monitored in patients receiving treatment (AIII).
Adverse effects of amphotericin B are primarily nephrotoxicity; permanent nephrotoxicity is related to cumulative dose. Infusion-related fevers, chills, nausea, and vomiting can occur, although they are less frequent in children than in adults. Renal dysfunction and electrolyte imbalances are its primary toxicities; these parameters should be monitored during therapy.
Itraconazole, like other azoles, has relatively low rates of toxicity. GI upset is seen occasionally and its principal toxicity is hepatic. The azole drugs inhibit CYP450-dependent hepatic enzymes so that drug interactions, particularly with antiretroviral drugs, should be carefully evaluated before initiation of therapy.
IRIS caused by an inflammatory response to histoplasmosis unmasked by HAART-induced improvement in cellular immunity is unusual, and symptoms are often mild (404). In the event of IRIS, antiretroviral therapy should be continued along with antifungal therapy (AIII).
Management of Treatment Failure
Both voriconazole and posaconazole have been used successfully in a small number of refractory cases in adults (382). Because little experience has been reported using the newer azoles and data are limited on use of these agents in children, expert consultation is recommended for cases refractory to first-line agents.
Prevention of Recurrence
Children responding well after completion of initial amphotericin B treatment should be continued on oral itraconazole maintenance therapy for at least 1 year (AII). Longer-term suppressive therapy with itraconazole may be required in HIV-infected children who are severely immunosuppressed (i.e., CD4 <15% or <150 cells/mm3 in children aged ≥6 years) and patients who experience relapse despite receipt of appropriate therapy (AII) (382,398). Fluconazole is less effective than itraconazole (CII), and experience is limited in children with voriconazole.
Discontinuing Secondary Prophylaxis
Discontinuation of treatment has not been examined in children. Data from a clinical trial evaluating discontinuation in adults with immune restoration on HAART suggest discontinuation of itraconazole is recommended in adults if itraconazole has been received for ≥1 year, blood cultures are negative, histoplasmal serum antigen is <2 ng/mL, CD4 counts are >150 cells/mm3, and the patient is compliant with HAART (AI) (398). Extrapolating these recommendations to HIV-infected children on HAART with immune restoration (i.e., CD4 >150 cells/mm3 in children aged ≥6 years) seems reasonable (CIII). Treatment should resume if these parameters are not met. Chronic suppressive therapy is recommended for relapse that occurs despite appropriate treatment (BIII).
Pneumocystis Pneumonia
Epidemiology
Pneumocystis spp. are found worldwide in the lungs of humans and lower animals. The organisms are host specific, and cross-infection between humans and other animals does not occur. Pneumocystis spp. from all sources are morphologically, tinctorially, and biologically similar, but surface antigens and gene sequencing have demonstrated host-specific differences. Since the original designation of Pneumocystis carinii a century ago, several changes in terminology have been suggested. The most recent proposal to change P. carinii to P. jirovecii for isolates from human lungs has gained some popularity but remains controversial.
Pneumocystis has been designated a fungus on the basis of DNA analysis, but it has several biologic features of protozoa. It is one of the most frequent causes of infection in humans. By age 2--4 years, more than 80% of children in most countries have acquired antibodies to Pneumocystis (405--407). Immunocompetent infants with the infection are either asymptomatic or have mild respiratory symptoms. Pneumocystis pneumonia (PCP) occurs almost exclusively in the immunocompromised host.
PCP remains a common AIDS-indicator disease among HIV-infected children. The highest incidence of PCP in HIV-infected children is in the first year of life, with cases peaking at age 3--6 months (408--410). Data from the CDC Pediatric Spectrum of Disease Project (1994--2001) indicate a decline in PCP infection rates (cases per 1000 HIV-infected children) from 25 in 1994 to 18 in 1996 to six in 2001 (1,411). Similarly, analyses of data from the Perinatal AIDS Collaborative Transmission Study revealed a 95% decline in PCP (cases per 100 child-years) from 5.8 (pre-HAART era) to 0.3 (HAART era) (4). Finally, the incidence rate of PCP (cases per 100 child-years) was 1.3 during the pre-HAART era (1981--1988) and <0.5 during the HAART era (2001--2004) (3). This decline probably resulted from implementation of interventions to prevent mother-to-child transmission of HIV, introduction of HAART in HIV-infected children in 1995, and chemoprophylaxis for PCP.
PCP is a major cause of death among HIV-infected infants and children in Africa. Autopsies revealed PCP in 16% of children dying with HIV/AIDS during 1992 and 1993 (412), in 29% of those dying during 1997 and 2000 (413), and in 44% of those dying during 2000 and 2001 (414).
The mode of transmission of Pneumocystis among HIV-infected infants, children, and adults is not firmly established, but airborne human-to-human transmission is likely. Animal studies show Pneumocystis is transmitted by air from infected to susceptible rats (415,416). Human-to-human transmission has been suggested by molecular epidemiology and global clustering of PCP cases in recent studies (417--419). Intrauterine transmission is considered rare. However, in one report, one of eight infants born to women who had AIDS and PCP during pregnancy had evidence of Pneumocystis infection (420).
The single most important factor in susceptibility of HIV-infected patients of all ages to PCP is the status of cell-mediated immunity of the host. Severe compromise, reflected by a marked decrease in CD4 count and percentage, is the hallmark of high risk for PCP and is discussed further in the prevention section.
Clinical Manifestations
Prominent clinical features of PCP among HIV-infected children are fever, tachypnea, dyspnea, and cough. The severity of these signs and symptoms varies from child to child. Onset can be abrupt or insidious with nonspecific symptoms (e.g., mild cough, dyspnea, poor feeding, diarrhea, and weight loss). Some patients may not be febrile, but almost all will have tachypnea by the time pneumonitis is evident on chest radiograph. Physical examination sometimes shows bilateral basilar rales with evidence of respiratory distress and hypoxia.
In HIV-infected children with pneumonia, four clinical variables are independently associated with PCP: age <6 months, respiratory rate >59 breaths per minute, arterial percentage hemoglobin saturation ≤92%, and the absence of vomiting (421). A high plasma HIV RNA concentration strongly predicts PCP and other OIs (422).
Extrapulmonary Pneumocystis organisms are found in <2.5% of HIV-infected adults and children (423,424). This can occur without concurrent PCP and can be located at multiple noncontiguous sites. Involved sites have included ear, eye, thyroid, spleen, GI tract, peritoneum, stomach, duodenum, small intestine, transverse colon, liver, and pancreas. Less frequently involved sites include adrenal glands, muscle, bone marrow, heart, kidney, ureter, lymph nodes, meninges, and cerebral cortex.
Diagnosis
Most children with PCP have substantial hypoxia with low arterial oxygen pressure and a PaO2 >30 mm Hg. The CD4 count is often <200 cells/mm3 and the CD4 percentage <15% in children aged >5 years. Lactic dehydrogenase is often increased, but this is not specific for PCP. Serum albumin may be depressed. Chest radiographs most commonly indicate bilateral diffuse parenchymal infiltrates with "ground-glass" or reticulogranular appearance, but they also can be normal or have only mild parenchymal infiltrates. The earliest infiltrates are perihilar, progressing peripherally before reaching the apical portions of the lung. Rarely, lobar, cavitary, nodular, or miliary lesions; pneumothorax; or pneumomediastinum are observed.
A definitive diagnosis of PCP requires demonstration of the organism in pulmonary tissues or fluids in the presence of pneumonitis. Diagnostic procedures are the same as for adults suspected to have PCP (see Guidelines for the Prevention and Treatment of Opportunistic Infections in HIV-Infected Adults) (16), but some procedures may be more difficult to perform in children. Several procedures for collecting and staining specimens are available.
Induced sputum analysis, during which the patient produces sputum after inhalation of nebulized 3% hypertonic saline, may be difficult among children aged <2 years because of small airways and poor ability to produce sputum. Complications from the procedure include nausea, vomiting, and bronchospasm. Sensitivity of sputum analysis in adults ranges from 25% to 90%. After a negative induced sputum sample, a bronchoalveolar lavage for definitive diagnosis may be necessary.
Nasogastric aspirates, if positive, are of diagnostic value. Pneumocystis organisms were found in 48.6% of HIV-infected children with respiratory illnesses in whom gastric aspirates were obtained on three consecutive mornings (425). Other studies have shown the organism is not found in gastric contents without PCP (426).
Bronchoscopy with bronchoalveolar lavage is the diagnostic procedure of choice for most infants and children. Sensitivity ranges from 55% to 97% and may be positive for ≥72 hours after initiation of PCP treatment; treatment should not be delayed while awaiting results. Complications include hemoptysis, pneumothorax, transient increase in hypoxemia, a transient increase in pulmonary infiltrates at the lavage site, and postbronchoscopy fever.
Fiberoptic bronchoscopy with transbronchial biopsy is recommended only when bronchoalveolar lavage is negative or nondiagnostic despite a clinical picture consistent with PCP. Sensitivity is 87%--95%, and cysts can be identified up to 10 days after initiation of treatment (up to 4--6 weeks in certain patients). Complications include pneumothorax and hemorrhage; this procedure is contraindicated in children with thrombocytopenia.
Open-lung biopsy is the most sensitive and specific diagnostic technique, but because it requires thoracotomy and often chest tube drainage, is not recommended routinely. It has the advantage of revealing the type and extent of disease as well as the organism. Histopathology shows alveoli filled with eosinophilic, acellular, proteinaceous material that contains cysts and trophozoites but few inflammatory cells. Complications include pneumothorax, pneumomediastinum, and hemorrhage.
Three types of stains can be used to identify Pneumocystis organisms in specimens. Gomori methenamine-silver method stains the cyst wall brown or black. Toluidine blue stains the cyst wall blue or lavender. Both methods stain fungal elements. Giemsa, Diff-Quick(r), and Wright stains depict the trophozoites and intracystic sporozoites pale blue with a punctate red nucleus, but unlike other stains, these do not stain the cyst wall. Monoclonal immunofluorescent antibodies (Merifluor(r), Meridian Bioscience, Inc., Cincinnati, OH) that stain the cyst wall also can be used for diagnosis and have enhanced specificity compared with the other methods. A cyst wall, trophozoite, and immunofluorescent antibody stain is recommended for each specimen studied.
PCR assays to amplify the human Pneumocystis MSG gene, mitochondrial large subunit (mtlsu) rRNA, the dihydropteroate synthase gene, and the internal transcribed spacer region genes have been developed for diagnostic evaluation. These tests are usually more sensitive but less specific than microscopic methods and are not standardized or available in most centers (427,428). Pneumocystis-specific DNA is found in 18% of bronchoalveolar lavage samples from patients without clinical PCP, HIV, and other infections (429).
Coinfection with other organisms (e.g., CMV or pneumococcus) has been reported in HIV-infected children (410,430,431). Children with dual infections may have more severe disease. Although CMV in lung secretions of children with PCP indicates colonization, it usually does not require therapy. The presence of Pneumocystis is always an indication for treatment.
Prevention Recommendations
Preventing Exposure
The need for isolation of hospitalized persons who have PCP has been neither demonstrated nor discounted. PCP patients clearly do not need to be isolated from persons with normal immune responses and from immunocompromised high-risk patients who are receiving PCP prophylaxis. Under unusual circumstances where prophylaxis cannot be administered, avoid placement in the same room with an immucompromised patient (CIII).
Preventing First Episode of Disease
Chemoprophylaxis is highly effective in preventing PCP. Criteria for its use are based on the patient's age and CD4 count or percentage (AII). Prophylaxis is recommended for all HIV-infected children aged ≥6 years who have CD4 counts <200 cells/mm3 or CD4 <15%, for children aged 1--5 years with CD4 counts of <500 cells/mm3 or CD4 <15%, and for all HIV-infected infants aged <12 months regardless of CD4 count or percentage (432).
Infants born to HIV-infected mothers should be considered for prophylaxis beginning at 4--6 weeks of age. HIV-infected infants should be administered prophylaxis until 1 year of age, at which time they should be reassessed on the basis of the age-specific CD4 count or percentage thresholds mentioned above (AII). Infants with indeterminate HIV infection status should receive prophylaxis until they are determined to be HIV-uninfected or presumptively uninfected with HIV (AIII). Prophylaxis is not recommended for infants who meet criteria for definitively or presumptively HIV-uninfected. In nonbreast-feeding infants with no positive HIV virologic test results, presumptive exclusion of HIV infection can be based on two negative virologic test results: one obtained at ≥2 weeks and one obtained at ≥4 weeks of age, one negative virologic test result obtained at ≥8 weeks of age, or one negative HIV-antibody test result obtained at ≥6 months of age. Definitive exclusion of HIV infection is based on two negative virologic test results: one obtained at ≥1 month of age and one obtained at ≥4 months of age, or on two negative HIV-antibody test results from separate specimens obtained at ≥6 months of age. For both presumptive and definitive exclusion of infection, the child should have no other laboratory (e.g., no positive virologic test results) or clinical (e.g., no AIDS-defining conditions) evidence of HIV infection.
Four drug regimens have been found effective and relatively safe for preventing PCP in high-risk HIV-infected children and adults.
Trimethoprim--sulfamethoxazole (TMP--SMX) (Cotrimoxazole) is the drug of choice for prophylaxis because of its high efficacy, relative safety, low cost, and broad antimicrobial spectrum (AI). TMP alone has little, if any, anti-Pneumocystis activity, but it enhances the activity of the sulfonamide. The prophylactic dosage is 150 mg/m2 body surface area/day TMP and 750 mg/m2 body surface area/day SMX (approximately 5.0 mg/kg/day TMP and 25 mg/kg/day SMX) administered orally in equally divided doses twice a day on 3 consecutive days per week (433). The total daily dose should not exceed 320 mg TMP and 1600 mg SMX. For patients with impaired renal function, a reduced dose may be necessary.
Alternatively, TMP--SMX can be administered daily 7 days a week (AI) (434). TMP--SMX also is effective in preventing toxoplasmosis (435) and some bacterial infections (e.g., Salmonella, Haemophilus, Staphylococcus) (434,436--438).
Dihydropteroate synthase gene mutations in Pneumocystis from humans have been observed with TMP--SMX and dapsone prophylaxis, suggestive of possible drug resistance, but studies for clinical correlates have not provided conclusive results (427). More apparent is the association of prolonged TMP--SMX prophylaxis for PCP with the emergence of selective pressure resistance of clinically important bacterial species to TMP-SMX, a point to be considered in managing bacterial infections in patients receiving prophylaxis (439,440).
Other effective and safe prophylaxis regimens are available for patients unable to take TMP-SMX. A second choice would be either atovaquone or dapsone (BI) (441). Atovaquone is effective and safe but expensive. Dapsone is effective and inexpensive but associated with more serious adverse effects than atovaquone.
Atovaquone is administered with a meal as an oral yellow suspension in single dosage of 30 mg/kg/day for patients 1--3 months and >24 months of age, and 45 mg/kg/day for infants aged 4--24 months (442). Unlike TMP-SMX, atovaquone has no antibacterial activity but is effective against Toxoplasma gondii. Azithromycin, in a single dosage of 5.0 mg/kg/day has been used to supplement atovaquone for greater broad-spectrum prophylaxis. The randomized, double-blind, placebo-controlled study PACTG 254 compared TMP-SMX and atovaquone plus azithromycin for 3 years (median) in 366 HIV-infected children qualifying for PCP prophylaxis (95). Results showed atovaquone-azithromycin to be as effective as TMP-SMX for preventing serious bacterial infections, as well as PCP.
Dapsone can be administered on a daily or weekly schedule as 2.0 mg/kg/day (maximum total dosage of 100 mg/day) or 4.0 mg/kg/week (maximum total dosage of 200 mg/week) orally. Approximately two thirds of patients intolerant to TMP-SMX can take dapsone successfully. Studies in adults show dapsone is as effective as atovaquone or aerosolized pentamidine but slightly less effective than TMP-SMX (441,442).
Aerosolized pentamidine is recommended for children who cannot take TMP-SMX, atovaquone, or dapsone and are old enough to use nebulization with a Respirgard II(r) nebulizer (Marquest, Englewood, CO) (BI). The dosage for all ages is 300 mg once a month (437). Adverse reactions among HIV-infected children include cough, sneezing, and bronchospasm (443).
Pyrimethamine-sulfadoxine (Fansidar(r)) also is recognized as an effective prophylaxis regimen in adults (CIII). Although this drug was effective in preventing PCP in Iranian orphanages in the 1960s, it has not been evaluated adequately among HIV-infected children.
The use of IV pentamidine is not recommended for prophylaxis (EIII) (444).
Discontinuing Primary Prophylaxis
Studies of HIV-infected adults and children following immune reconstitution after receipt of HAART demonstrate acceptable low risks for PCP after discontinuation of prophylaxis (46,445--449). Data from the PACTG 1008 study evaluated 235 HIV-infected children and adolescents on antiretroviral therapy who received PCP prophylaxis ≥6 months and achieved CD4 percentages of ≥20% for patients aged >6 years and ≥25% for patients aged 2--6 years, after which the prophylaxis was stopped (46). During the median follow-up of 2.5 years (547 person-years), no cases of PCP occurred; 9.4% of patients enrolled required reinstitution of PCP prophylaxis during observation. These data, along with data from studies in adults, support the expectation for very low risk for PCP after prophylaxis is discontinued in children who have achieved immune reconstitution.
Discontinuation of PCP prophylaxis should be considered for HIV-infected children when, after receiving HAART for ≥6 months, CD4 percentage is ≥15% or CD4 count is ≥200 cells/mm3 for patients aged >6 years (BII) and CD4 percentage is ≥15% or CD4 count is ≥500 cells/mm3 for patients aged 1--5 years (BII) for >3 consecutive months. Subsequently, the CD4 percentage and count should be reevaluated at least every 3 months and prophylaxis reinstituted if the original criteria for prophylaxis are reached (BIII). PCP prophylaxis is not to be discontinued in HIV-infected infants aged <1 year.
Treatment Recommendations
Treatment of Disease
TMP-SMX is the recommended treatment for PCP (AI). The dose for HIV-infected children aged >2 months is 15--20 mg/kg/day of the TMP component and 75--100 mg/kg/day of the SMX component administered intravenously in 3 or 4 divided doses, with the dose infused over 1 hour for 21 days (AI). As the acute pneumonitis subsides, children with mild to moderate disease who do not have malabsorption or diarrhea can be administered oral treatment with the same dose of TMP-SMX in 3 or 4 divided doses to complete a 21-day course (AII). Effective therapeutic serum concentrations of 5--10 µg/mL TMP can be reached with the recommended dose administered orally in HIV-infected children (450).
IV pentamidine isethionate once daily is recommended for patients who cannot tolerate TMP-SMX or who demonstrate clinical treatment failure after 5--7 days of TMP-SMX therapy (AI). No evidence exists for synergistic or additive effects on efficacy of these agents; therefore, because of potential increased toxicity, their combined use is not recommended (DIII). Among patients with clinical improvement after 7--10 days of IV therapy with pentamidine, an oral regimen (e.g., atovaquone or TMP/dapsone) might be considered to complete a 21-day course (BIII).
Atovaquone is an alternative for treatment of mild to moderately severe PCP in adults (BI) (95). Therapeutic data are limited for children, but the dosage of 30--40 mg/kg/day in two divided doses administered orally is established for children <3 months and ≥24 months of age. Children aged 3--24 months require a higher dosage of 45 mg/kg/day (AII) (442). The dosage for adolescents and adults is 750 mg twice daily. Food increases the bioavailability of atovaquone approximately threefold compared with that achieved with the fasting state. Atovaquone concentration increases with coadministration of fluconazole and prednisone and decreased by coadministration with acyclovir, opiates, cephalosporins, rifampin, and benzodiazepines.
Dapsone/TMP is effective in treating mild to moderate PCP among adults (BI) (451); data on toxicity and efficacy among children are limited. The dosage of dapsone for adolescents and adults is 100 mg (total dose) orally once daily and TMP 15 mg/kg/day divided into three daily doses administered for 21 days. Among children aged <13 years, a dapsone dosage of 2 mg/kg/day is required to achieve therapeutic levels in children (AII) (452). The pediatric dose of TMP is 15 mg/kg/day divided into three daily doses. Dapsone is less effective than the combination (453).
Clindamycin/primaquine has been used to treat mild to moderate PCP among adults (BI); data for children are not available (CIII). Primaquine is contraindicated for patients with glucose-6-dehydrogenase deficiency because of the possibility of inducing hemolytic anemia. Dose information for treating PCP is available only for adults. For patients weighing >60 kg, clindamycin 600 mg intravenously every 6 hours for 10 days, then 300--450 mg orally every 6 hours to complete 21 days of treatment is recommended. Primaquine is administered as 30 mg of base orally for 21 days. Dosing for children is based on use of these drugs for treating other infections: the usual pediatric dose of clindamycin for treating bacterial infection is 10 mg/kg/dose every 6 hours, and the pediatric dose of primaquine equivalent to an adult dose of 20 mg base (when used for malaria) is 0.3 mg/kg/day of the base.
On the basis of studies in adults, a short course of corticosteroids is recommended in cases of moderate or severe PCP, starting within 72 hours of diagnosis (AI). Pediatric studies have indicated reduced acute respiratory failure, decreased need for ventilation, and decreased mortality with early use of corticosteroids in HIV-infected children who have PCP (454--456). Indications for corticosteroid treatment include a PaO2 value of <70 mm Hg or an alveolar-arterial gradient of >35 mm Hg. Doses for children vary between studies. A commonly used scheme is prednisone on days 1--5, 1 mg/kg/dose twice daily; days 6--10, 0.5 mg/kg/dose twice daily; and days 11--21, 0.5 mg/kg once daily. Alternative regimens include 1) adult dosage prednisone on days 1--5, 40 mg twice daily; days 6--10, 40 mg once daily; days 11--21, 20 mg once daily; and 2) methylprednisolone (IV) on days 1--7, 1 mg/kg/dose every 6 hours; days 8--9, 1 mg/kg/dose twice daily; days 10 and 11, 0.5 mg/kg/dose twice daily; days 12--16, 1 mg/kg once daily.
Some case reports have documented improved pulmonary function with use of surfactant in cases of severe disease (e.g., respiratory distress syndrome with established respiratory failure requiring ventilation) (457--459). Alterations in surfactant function and composition have been demonstrated in HIV-infected adults with PCP (460). Data are insufficient to recommend surfactant administration for PCP.
Monitoring and Adverse Events, Including IRIS
Clinical parameters to monitor the status of disease include temperature, respiratory rate, arterial oxygen saturation, and chest radiograph (461). Clinical improvement can be expected at a mean of approximately 4.5 ± 2.5 days and radiographic improvement at approximately 7.7 ± 4.5 days (461).
IRIS has been less frequently associated with Pneumocystis infection (2% of 44 adults with IRIS) than with several other OIs in HIV-infected adults and children (462). Whether this low rate is related to PCP prophylaxis is not known.
In children, adverse reactions to TMP-SMX include rash (mild maculopapular in most cases but rarely erythema multiforme and Stevens-Johnson syndrome), hematologic abnormalities (e.g., neutropenia, thrombocytopenia, megaloblastic or aplastic anemia), GI complaints (usually mild), hepatitis, and renal disorders (e.g., interstitial nephritis) (463,464). Data from a PACTG study of HIV-infected children at high risk for PCP receiving TMP-SMX for a median of 3 years showed 28% had a rash, 9.3% had neutropenia, 8.8% had thrombocytopenia, and 2.2% had anemia (95). None were fatal or nonreversible reactions. Some very mild reactions will resolve while the drug is continued. With any significant adverse effect, TMP-SMX should be withheld until the reaction has subsided. Unless the reaction has been life-threatening, TMP-SMX prophylaxis can be resumed, preferably by beginning with low desensitizing daily doses and gradually increasing to full doses (BII) (465,466). In adults, 75% of patients affected tolerate rechallenge with TMP-SMX (466). The overall frequency of adverse reactions appears to be lower among HIV-infected children than among adults; approximately 15% of children have substantial adverse reactions to TMP-SMX (446). If an urticarial rash or Stevens-Johnson syndrome occurs, TMP-SMX should be discontinued and not readministered (EIII).
The most common adverse drug reaction to pentamidine isethionate is renal toxicity, which usually occurs after 2 weeks of therapy and can be averted by adequate hydration and careful monitoring of renal function and electrolytes. Severe hypotension (particularly if infused rapidly), prolonged QT interval (torsades de pointes), and cardiac arrhythmias can occur. Hypoglycemia (usually after 5--7 days of therapy) or hyperglycemia, hypercalcemia, hyperkalemia, pancreatitis, and insulin-dependent diabetes mellitus also have been reported. The patient may experience a metallic or bitter taste. Serious adverse reactions to pentamidine have been reported in approximately 17% of children receiving the drug (467). This drug should not be administered with other nephrotoxic drugs (e.g., aminoglycosides, amphotericin B, cisplatin, or vancomycin) and with agents associated with pancreatitis (e.g., didanosine).
With dapsone and TMP, the primary adverse reaction is reversible neutropenia; other reactions include skin rashes, elevated serum transaminases, methemaglobinemia, anemia, and thrombocytopenia (451,453). Dapsone is the problematic component of the combination and accounts for most of the adverse reactions (442).
Skin rashes (10%--15%), nausea, and diarrhea can occur with atovaquone administration. Liver enzymes may increase briefly. No serious toxicity or fatality has been demonstrated from the use of atovaquone in adults or children.
Adverse reactions to clindamycin/primaquine include skin rash, nausea, and diarrhea.
Management of Treatment Failure
An inflammatory reaction to antibiotic-induced killing of the organism in the lungs can result in an initial early and reversible deterioration during the first 3--5 days of therapy, so an adequate trial of therapy is needed before switching drugs because of lack of clinical improvement. Clinical failure is defined by lack of improvement or worsening of respiratory function documented by arterial blood gases after at least 4--8 days of anti-PCP treatment. Other concomitant infections need to be excluded as causees of clinical failure. With evidence of treatment failure after the use of TMP-SMX, drugs can be changed. If tolerated, pentamidine isethionate is the drug of next choice (BII). No evidence exists for synergistic or additive therapeutic effects; therefore, because of potential increased toxicity, their combination is not recommended.
Prevention of Recurrence
None of the drugs administered to treat and prevent PCP completely eliminates Pneumocystis, and prophylaxis is effective only while the selected drug is administered. Patients who have experienced an episode of PCP should have prophylaxis administered continuously after completion of treatment (AI).
Discontinuing Secondary Prophylaxis
In most patients, secondary prophylaxis can be discontinued using the same criteria as for discontinuing primary prophylaxis. PCP prophylaxis is not to be discontinued in HIV-infected infants <1 year. Children who present with clinical signs and symptoms compatible with PCP after discontinuation of prophylaxis should be evaluated thoroughly despite normal or high CD4 counts or percentages (BII) (468).
Parasitic Infections
Cryptosporidiosis/Microsporidiosis
Epidemiology
Cryptosporidium spp. are protozoal parasites that mainly cause enteric illness (e.g., chronic diarrhea) in humans and animals; the parasites have worldwide distribution. The three most common species infecting humans are C. hominis, C. parvus, and C. meleagridis. In addition, infections with C. canis, C. felis, C. muris, and Cryptosporidium pig genotype have been reported in immunocompromised patients. Cryptosporidium parasites usually invade the small bowel, but in immunocompromised hosts, the large bowel and extraintestinal sites also are involved.
The parasite is transmitted by ingestion of oocysts excreted in the feces of infected animals and humans. The parasite is highly infectious, with an ID50 ranging from nine to 1042 oocysts, depending on the isolate (469). Infection occurs when the ingested oocyst releases sporozoites, which attach to and invade the intestinal epithelial cells. The parasite has a predilection for the jejunum and terminal ileum (469).
Person-to-person transmission is common in child care centers; infants with cryptosporidiosis-associated diarrhea can infect adults during diapering (470). Oocysts can contaminate recreational water sources (e.g., swimming pools, lakes) and public water supplies and may persist despite standard chlorination. Physical steps---flocculation, sedimentation, or filtration---are necessary to remove the parasite from the water. Outbreaks have been associated with ingestion of contaminated drinking water in large metropolitan areas that have chlorination but not filtration systems and with public swimming pools (471). Foodborne and person-to-person spread have been documented (469).
Cryptosporidiosis also occurs among international travelers. Although fewer than 4000 such cases were reported each year in the United States during 1995--2002, an estimated 300,000 persons are infected each year; underuse of diagnostic tests and poor sensitivity of the older tests combined with underreporting are the main reasons for this difference (472). In industrialized countries, the prevalence of cryptosporidiosis among children is usually considered to range from 3.0% to 3.6% (473); it is reported more frequently among children in developing countries (474--476).
Before the advent of effective antiretroviral therapy, cryptosporidiosis was diagnosed primarily in patients with advanced HIV disease and AIDS. However, the incidence has declined dramatically in areas where HAART became widely available (476,477).
Microspora spp. are obligate, intracellular, spore-forming protozoa that primarily cause moderate to severe diarrhea among children. They are related to fungi but defined by their unique single polar tube that coils around the interior of the spore (478). Many microsporidia were reported as pathogens in humans, but Enterocytozoon bieneusi and Encephalitozoon intestinalis are the most common microsporidia that cause infection among patients with HIV infection. Other microsporidia such as Encephalitozoon cuniculi, Encephalitozoon hellem, Trachipleistophora hominis, T. anthropophthera, Pleistophora spp., P. ronneeafiei, Vittaforma (Nosema) corneae, Microsporidium spp., Nosema ocularum, Anncaliia (syns Brachiola/Nosema) connori, A. (syn Brachiola) vesicularum, and A. (syns Brachiola/Nosema) algerae also have been implicated in human infections. The Microspora parasites develop in enterocytes and are excreted with feces and, like C. parvum, are transmitted by the fecal-oral route, including through ingestion of contaminated food or water (479). They cause infection in HIV-uninfected children, elderly persons, travelers, and organ transplant recipients. Although the incidence of microsporidiosis has declined dramatically in areas where HAART is used, it continues to affect HIV-infected patients who are unable to receive or continue HAART.
Clinical Manifestations
Symptoms of cryptosporidiosis develop after an incubation period of 1 week. Frequent, usually nonbloody, persistent watery diarrhea is the most common manifestation of both cryptosporidial and microsporidial infection, with abdominal cramps, fatigue, vomiting, anorexia, weight loss, and poor weight gain. Fever and vomiting are relatively common in children, mimicking viral gastroenteritis (474). In almost half of the cases, the diarrhea persists for >2 weeks (480). Longer episodes of diarrhea have been associated with increased risk for recurrent episodes and weight loss (481). Among immunocompromised children, chronic severe diarrhea can result in malnutrition, failure to thrive, and substantial intestinal fluid losses, resulting in severe dehydration and even death. Clinical history or physical examination does not allow differentiation of cryptosporidial or microsporidial disease from those caused by other pathogens.
Cryptosporidium can migrate into the bile duct and result in inflammation of the biliary epithelium, causing acalculous cholecystitis, and sclerosing cholangitis (482). Symptoms and signs include fever, right upper abdominal pain, and elevated alkaline phosphatase. Pancreatitis occurs rarely. Although infection usually is limited to the GI tract, pulmonary or disseminated infection can occur among immunocompromised children.
The most common manifestation of microsporidiosis is GI tract infection. In addition to the more common acute and chronic diarrhea, microsporidia species have been described as causing hepatitis, peritonitis, keratoconjunctivitis, myositis, cholangitis, respiratory disease, sinusitis, encephalitis, and disseminated disease (483). Different infecting species may result in different clinical syndromes. Enterocytozoon bieneusi is associated with malabsorption, diarrhea, and cholangitis. Encephalitozoon cuniculi is associated with hepatitis, encephalitis, and disseminated disease. Encephalitozoon (syn Septata) intestinalis is associated with diarrhea, disseminated infection, and superficial keratoconjuctivitis. Encephalitozoon hellem is associated with superficial keratoconjunctivitis, sinusitis, respiratory disease, prostatic abscesses, and disseminated infection. Nosema, Vittaforma, and Microsporidium are associated with stromal keratitis following trauma in immunocompetent hosts. Pleistophora, Anncaliia, and Trachipleistophora are associated with myositis. Trachipleistophora is associated with encephalitis and disseminated disease.
Diagnosis
Because Cryptosporidium cannot be grown on laboratory media, cryptosporidiosis usually is diagnosed by microscopic identification of the oocysts in stool or tissue. Stool samples are concentrated using the sucrose flotation or formalin-ethyl acetate methods. Monoclonal antibody-based fluorescein-conjugated stain for oocysts and an EIA to detect antigen in stool are preferred to staining methods (e.g., with a modified Kinyoun acid-fast stain) because of enhanced sensitivity and specificity (484,485). Antigen-detection assays (e.g., enzyme-linked immunosorbent assay, immunochromatography) are available commercially that have good sensitivity and excellent specificity (485,486). Molecular methods such as PCR hold promise to further enhance sensitivity (487). Among persons with profuse diarrheal illness, one stool specimen is usually adequate for diagnosis. However, oocyst excretion can be intermittent; therefore, the parasite might not be detected in every stool, and among persons with milder disease, repeat stool sampling is recommended. Organisms also can be identified on small intestinal biopsy or intestinal fluid samples.
To diagnose microsporidia infection, thin smears of unconcentrated stool-formalin suspension or duodenal aspirates can be stained with modified trichrome stain. Chemofluorescent agents such as chromotrope 2R and calcofluor white (a fluorescent brightener) are useful as selective stains for microsporidia in stool and other body fluids. Microsporidia spores are small (1--5 µm diameter), ovoid, stain pink to red with modified trichrome stain, and contain a distinctive equatorial-belt-like stripe.
Urine sediment examination by light microscopy can be used to identify microsporidia spores causing disseminated disease (e.g., Encephalitozoonidae, Trachipleistophora). Transmission electron microscopy or PCR (using specific primers) is needed for speciation.
Endoscopic biopsy should be considered for all patients with chronic diarrhea of >2 months' duration and negative stool examinations. Touch preparations are useful for rapid diagnosis (i.e., within 24 hours). Sensitive assays using PCR amplification of parasite DNA sequences extracted from stool or biopsy specimens have been developed for Cryptosporidium and for Enterocytozoon bieneusi (487,488) but are research tools and not commercially available.
Prevention Recommendations
Preventing Exposure
Caregivers and HIV-infected children should be educated and counseled about the different ways Cryptosporidium can be transmitted. Modes of transmission include directly contacting fecal material from adults, diaper-aged children, and infected animals; contacting contaminated water during recreational activities; drinking contaminated water; and eating contaminated food.
Hand washing after exposure to potentially fecally contaminated material, including diapers, is important in reducing the risk for Cryptosporidium infection. HIV-infected children should not be allowed contact with ill pets or stool from pets, particularly dogs and cats <6 months of age; stray pets; or surfaces contaminated with human or animal stool. HIV-infected children should avoid direct contact with calves and lambs at farms or petting zoos.
HIV-infected children should not be allowed to drink water directly from lakes or rivers, including swallowing water while swimming or playing in recreational water. Caregivers and HIV-infected children should be aware that lakes, rivers, salt-water beaches, certain swimming pools, recreational water parks, and ornamental water fountains might be contaminated with human or animal waste that contains Cryptosporidium.
Some outbreaks of cryptosporidiosis have been linked to ingestion of water from municipal water supplies. During outbreaks or in other situations in which a community advisory to boil water is issued, water used in preparing infant formula and for drinking should be boiled for ≥3 minutes to eliminate risk for cryptosporidiosis.
Nationally distributed brands of bottled or canned carbonated soft drinks are safe to drink. Commercially packaged noncarbonated soft drinks and fruit juices that do not require refrigeration until after they are opened (i.e., can be stored unrefrigerated on grocery shelves) also are safe. Nationally distributed brands of frozen fruit juice concentrate are safe if they are reconstituted by the user with water from a safe water source. Fruit juices that must be kept refrigerated from the time they are processed to the time of consumption might be either fresh (i.e., unpasteurized) or heat-treated (i.e., pasteurized); only juices labeled as pasteurized should be considered free of risk from Cryptosporidium. Other pasteurized beverages also are considered safe to drink.
Cryptosporidium-infected patients should not work as food handlers, especially if the food to be handled is intended to be eaten without cooking.
In a hospital, standard precautions (i.e., use of gloves and hand washing after removal of gloves) should be sufficient to prevent transmission of cryptosporidiosis from an infected patient to a susceptible HIV-infected person. However, because of the potential for fomite transmission, some experts recommend that severely immunocompromised HIV-infected patients not share a room with a patient with cryptosporidiosis (CIII).
Similar to precautions for preventing cryptosporidiosis, attention to hand washing and other personal hygiene measures will reduce exposure to microsporidia.
Preventing First Episode of Disease
Because chronic Cryptosporidium infection occurs most frequently in HIV-infected persons with advanced immune deficiency, antiretroviral treatment of HIV-infected children before development of severe immune deficiency is a primary modality of prevention (AII).
Some observational studies from the pre-HAART era suggested that rifabutin or clarithromycin prophylaxis for MAC may be associated with decreased rates of cryptosporidiosis (489,490). However, data are conflicting and insufficient to recommend using these drugs solely for prophylaxis of cryptosporidiosis. No chemoprophylactic regimens are known to be effective in preventing microsporidiosis.
Discontinuing Primary Prophylaxis
Not applicable.
Treatment Recommendations
Treatment of Disease
Immune reconstitution resulting from HAART frequently will result in clearance of Cryptosporidium and Microsporidium infections. Effective HAART is the primary initial treatment for these infections in HIV-infected children and adults (AII) (477,491). Supportive care with hydration, correction of electrolyte abnormalities, and nutritional supplementation should be provided (AIII). Antimotility agents should be used with caution among young children (CIII).
Cryptosporidium
No consistently effective therapy is available for cryptosporidiosis, and duration of treatment among HIV-infected persons is uncertain (492). Multiple agents have been investigated in small randomized controlled clinical trials of HIV-infected adults, including nitazoxanide, paromomycin, spiramycin, bovine hyperimmune colostrum, and bovine dialyzable leukocyte extract. No pharmacologic or immunologic therapy directed specifically against C. parvum has yet been shown consistently effective and durable when used alone without concomitant antiretroviral therapy (492).
A review of clinical trials of treatment for Cryptosporidia in immunocompromised patients, including those with HIV infection, found that no agent has proven efficacy for treating cryptosporidiosis in immunocompromised patients; however, in immunocompetent persons, nitazoxanide reduces the load of parasites. Given the seriousness of this infection among immunocompromised persons, use of nitazoxanide can be considered in immunocompromised HIV-infected children in conjunction with HAART for immune restoration (CIII) (492).
Nitazoxanide is approved in the United States to treat diarrhea caused by Cryptosporidium and Giardia lamblia among children and is available in liquid and tablet formulations (BI for HIV-uninfected and CIII for HIV-infected children). An Egyptian clinical trial among 100 HIV-uninfected adults and children randomized patients to a 3-day course of nitazoxanide or placebo (493). Nitazoxanide therapy reduced the duration of both diarrhea and oocyst shedding; among children, clinical response was 88% with nitazoxanide and 38% with placebo. No severe adverse events were reported, and adverse events that were reported were similar in the treatment and placebo groups in this study. A study in Zambia among 100 malnourished children (half of whom were HIV-infected) aged 12--35 months reported a clinical response in 56% of HIV-uninfected children treated with nitazoxanide compared with 23% receiving placebo (494). However, among the children with HIV infection, no benefit was observed from nitazoxinide (clinical response in 8% treated with nitazoxanide compared with 25% receiving placebo). These results may be due to the short course (3 days) of therapy as retreatment for additional 3 days increased the number of responders. In a study in HIV-infected adults who had CD4 counts >50 cells/mm3, 14 days of nitazoxanide resulted in 71% (10 of 14) response using 500 mg twice daily and 90% (9 of 10) using 1000 mg twice daily, compared with 25% with placebo (495). The recommended dose for children is 100 mg orally twice daily for children aged 1--3 years and 200 mg twice daily for children aged 4--11 years. A tablet preparation (500 mg twice daily) is available for children aged ≥12 years. All medications should be administered with food.
Paromomycin, a nonabsorbable aminoglycoside indicated for the treatment of intestinal amebiasis, is effective for treating cryptosporidiosis in animal models but is not specifically approved for treatment of cryptosporidiosis in humans. A review and a meta-analysis of the two randomized controlled trials comparing paromomycin with placebo among adults with AIDS found the drug was no more effective than placebo in reducing diarrheal frequency or parasite burden (492,496,497), and a clinical response to paromomycin is rare in patients with CD4 count <100 cells/mm3. Therefore, data do not support a recommendation for the use of paromomycin for cryptosporidiosis (DII).
Azithromycin has demonstrated some activity against C. parvum infection in a limited number of HIV-infected children (498). An azithromycin regimen of 10 mg/kg/day on day 1, and 5 mg/kg/day on days 2--10 rapidly resolved enteric symptoms in three of four HIV-infected children with cryptosporidiosis (498). However, data are insufficient to recommend use of this drug to treat cryptosporidial infection (CIII).
Microsporidium
Albendazole has activity against many species of microsporidia, but it is not effective against Enterocytozoon infections or V. corneae (499,500). Albendazole decreased diarrhea and sometimes eliminated the organism (498,500). Albendazole is recommended for initial therapy of intestinal and disseminated microsporidiosis caused by microsporidia other than Enterocytozoon bieneusi and V. corneae (AII).
Although two drugs, fumagillin and nitazoxanide, have been studied in small numbers of patients for treatment of Enterocytozoon bieneusi infection, neither has definitive evidence for efficacy in adequate and controlled trials. Fumagillin (Sanofi-Synthelabo Laboratories, Gentilly, France), a water-insoluble antibiotic made by Aspergillus fumigatus, and its synthetic analog TNP-470 (501) have each been used to treat microsporidiosis in animals and humans. In a placebo-controlled study of immunocompromised adults (including HIV-infected adults) with Enterocytozoon bieneusi microsporidiosis, fumagillin (20 mg/dose orally three times daily for 2 weeks) was associated with decreased diarrhea and clearance of microsporidial spores, which was not observed in placebo patients (501). No data are available on use of fumagillin or TNP-470 among HIV-infected children, and neither drug is available in the United States. Data are insufficient to make recommendations on the use of these drugs in children (CIII). One report indicated that treatment with nitazoxanide for 60 days might resolve chronic diarrhea caused by Enterocytozoon bieneusi in the absence of antiretroviral therapy (502), but this effect was minimal among patients with low CD4 counts and therefore may be of limited utility (CIII).
Keratoconjunctivitis caused by microsporidia among HIV-infected adults responds to topical therapy with investigational fumagillin eye drops prepared from Fumidil-B(r) (fumagillin bicylohexylammonium, a commercial product used to control a microsporidial disease of honeybees) in saline (to achieve a concentration of 70 µg/mL of fumagillin) (BII) (503). The combination of albendazole and fumagillin has demonstrated consistent activity against microsporidia in vitro and is recommended for ocular infections, in addition to topical therapy, because microsporidia may remain systemically despite clearance from the eye with topical therapy alone (BIII) (504).
Metronidazole and atovaquone are not active in vitro or in animal models and should not be used to treat microsporidiosis (EII).
Monitoring and Adverse Events, Including IRIS
Patients should be closely monitored for signs and symptoms of volume depletion, electrolyte and weight loss, and malnutrition. In severely ill patients, total parenteral nutrition may be indicated (BIII).
Nitazoxanide has not been associated with substantial side effects. Albendazole side effects are rare but hypersensitivity (e.g., rash, pruritis, fever), neutropenia (reversible), CNS effects (e.g., dizziness, headache), GI disturbances (e.g., abdominal pain, diarrhea, nausea, vomiting), hair loss (reversible), and elevated hepatic enzymes (reversible) have been reported. Dose-related bone marrow toxicity is the principal adverse effect of fumagillin, with reversible thrombocytopenia and neutropenia being the most frequent adverse events; topical fumagillin has not been associated with substantial side effects.
IRIS has not been described in association with treatment of cryptosporidiosis or with treatment for Enterocytozoon bieneusi or non-Enterocytozoon bieneusi microsporidiosis.
Management of Treatment Failure
The only feasible approaches to managing treatment failure are supportive treatment and optimization of ART to achieve full virologic suppression (AIII).
Prevention of Recurrence
No pharmacologic interventions are known to be effective in preventing recurrence of cryptosporidiosis or microsporidiosis. However, treatment for ocular microsporidiosis should be continued indefinitely because recurrence or relapse might follow treatment discontinuation (BIII).
Discontinuing Secondary Prophylaxis
Not applicable.
Malaria
Epidemiology
Malaria is an acute and chronic disease caused by obligate, intracellular protozoa of the genus Plasmodium. In 2005, CDC received reports of 1528 cases of malaria in the United States; of these, patient age was known for 1460, and 276 (18.9%) occurred in persons aged <18 years (505). For 220 pediatric patients aged <18 years for whom malarial species was known, most infections were caused by P. falciparum (70.9%) and P. vivax (19.5%) (505). Of 231 children for whom country of exposure was known, 76% of malarial infections were acquired in Africa, 16% in Asia and the Middle East, and 6.1% in the Americas (505).
Most (82%) malaria becomes symptomatic within 30 days after arrival in the United States; 99% of malaria cases become symptomatic within 1 year. Typically, 75% of U.S. travel-associated malaria occurs in persons who have not taken appropriate malaria chemoprophylaxis. Approximately 50% of infections among U.S. citizens are acquired during visits to friends and relatives in malaria-endemic regions (505). U.S.-born children of immigrants returning to visit family are at highest risk for malaria-associated death. In 2005, refugees or immigrants accounted for 28.3% of U.S. pediatric malaria cases, and the proportion of malaria among refugees and immigrants has not changed in the past decade (505--508).
No reliable data exist on the prevalence of HIV infection among immigrant and refugee children resettling in the United States because children aged <15 years are exempt from mandatory screening. Underlying HIV prevalence rates in children relocating to the United States are assumed to reflect the overall rates of HIV in the migrating population. For example, Somali refugees have very low rates of HIV, whereas rates in Central and West Africa are substantially higher. Presumably, children at risk for coinfection with HIV and malaria, particularly asymptomatic children or those with unrecognized disease, would be highest in populations where the underlying prevalence of both HIV and malaria are excessive (i.e., West, Central, and portions of East Africa).
Clinical Manifestations
In contrast to data suggesting increased frequency of both parasitemia and clinical malaria in HIV-infected adults (509,510), studies in young HIV-infected children in malaria-endemic areas usually have not found increased frequency or density of parasitemia with HIV coinfection (511--517), with one exception. Parasite density in children aged <5 years was higher in those with HIV infection than in those without HIV infection in Uganda (518). Additionally, most studies have not found malarial episodes to be more frequent or to present differently in HIV-infected than uninfected children (511,513). However, one study found that HIV-infected children aged >1 year may be more likely to have severe or complicated malaria (516). A study in Kenya suggested that HIV-exposed children aged <2 years (regardless of their infection status) were more likely than children without HIV exposure to have severe anemia with malarial episodes (517). Published studies on HIV/malaria coinfection have focused on adults and younger HIV-infected children; whether data from such studies applies to older HIV-infected children is uncertain. In a study in Uganda, HIV-infected persons aged ≥5 years with lower CD4 counts (<200 cells/mm3) had higher parasite counts during clinical malaria (518).
Signs and symptoms of malaria vary and depend on previous exposure (partial immunity), age of onset, and species of infecting organism but do not differ by HIV infection status in children. Fever is the most common symptom. In children, nonspecific symptoms predominate and may include chills, sweating, headache, myalgia, malaise, nausea, vomiting, diarrhea, and cough (519,520). These symptoms might increase the potential for misdiagnosis as a viral syndrome, upper respiratory tract infection, or gastroenteritis. Two thirds of all malaria in children in an area where malaria is nonendemic was misdiagnosed; children had two to four clinical visits before malaria was diagnosed (508).
Non-P. falciparum will be responsible for 75% of malaria diagnoses in nonimmune persons >30 days after they return to or travel to the United States from regions with malaria.
In contrast, the epidemiology in partially immune children may vary considerably. In one study, 60% of children migrating from malaria-holoendemic regions were smear-positive for P. falciparum 1 month after arrival in the United States (521). Since these data were collected, CDC has issued guidance to organizations resettling refugees to the United States to presumptively treat all refugees from sub-Saharan Africa, except under special circumstances, before they depart for the United States. This treatment should decrease malaria rates among refugees but does not completely eliminate risk because predeparture treatment with drugs effective against blood-stage parasites do not eliminate liver-stage parasites. However, nonrefugee immigrants from similar areas do not receive this presumptive therapy and are at greater risk for clinical manifestations of malaria after arrival in the United States. Therefore, children who have recently migrated from regions where malaria is highly endemic should be either presumptively treated for malaria or tested postarrival for malaria infection. CDC guidance can be found at http://www.cdc.gov/ncidod/dq/health.htm.
Chronic symptoms of splenomegaly, fever, and thrombocytopenia are highly specific for malaria in immigrant children and need appropriate evaluation (519,522). Congenital malaria is rare but should be considered in febrile neonates whose mothers migrated from areas where malaria is endemic; however, empiric therapy should not be administered without a diagnosis to febrile neonates of recent immigrants (519).
Diagnosis
Malaria is diagnosed by microscopic examination of a patient's blood. A Giemsa-stained thick blood smear is the most sensitive smear technique for detecting infection, with a thin blood smear used for accurate parasite speciation. In nonimmune persons, because symptoms may develop before parasitemia is detectable, several blood-smear examinations taken at 12- to 24-hour intervals may be needed to positively rule out malaria in symptomatic patients. The sensitivity and specificity of blood smear depends substantially on the laboratorian's skills. Assistance with microscopic diagnosis of malaria is available through state public health departments and the CDC Division of Parasitic Diseases diagnostic service (http://www.dpd.cdc.gov/dpdx).
Although blood smear examination remains the gold standard for laboratory confirmation of malaria, FDA has approved the Binax Now(r) Malaria Test, a rapid malaria antigen-capture assay, for use in clinical diagnosis of symptomatic malaria in children and adults. This test, which uses a monoclonal antibody to histidine-rich protein 2 to detect P. falciparum, and a monocloncal antibody aldolase to detect all malaria species, can be performed within 15 minutes. The studies presented to FDA show specificity approaching 100% for all malaria and a sensitivity of >95% for P. falciparum and 85%--95% for P. vivax. However, sensitivity for P. ovale, a less common form of human malaria, probably does not exceed 70%. Although this test performs well in symptomatic persons, preliminary data suggest poor performance of rapid diagnostic tests, including the Binax Now(r) Malaria Test for screening asymptomatic persons for malaria (523,524). Similarly, among asymptomatic immigrants, the sensitivity of a single blood smear is relatively poor (<50%) (523,524). Specific guidelines for laboratory diagnosis are summarized elsewhere and are available at the CDC malaria website (http://www.cdc.gov/malaria).
Prevention Recommendations
Preventing Exposure
HIV-infected children, particularly immunosuppressed children, should be advised to use personal protective measures to prevent mosquito bites when traveling to malaria-endemic areas (525). Specifically, clothing can be impregnated with permethrin, effective for weeks, even through 10 washings (AI). Long-acting DEET mosquito repellents (30% concentration) are practical and 99% effective when combined with permethrin-treated clothing (AI). DEET should be applied onto young children by caregivers and usually should not be applied to young children's hands (AIII). Specific instructions by providers on product purchasing and use are invaluable. Insecticide-treated bed nets are inexpensive and readily available in countries with endemic malaria. All children, regardless of their HIV infection status, should sleep under an insecticide-treated bed net when traveling in malaria-endemic areas.
Primary Prophylaxis
U.S.-born children of immigrant parents traveling to malaria-endemic regions are at especially high risk for malaria (525). Child immigrants, or second-generation immigrant children whose caregivers are from malaria-endemic areas, are likely to travel to high-risk destinations and be more susceptible than their caregivers because of lack of previous malaria exposure. These children and their caregivers are at especially high risk for acquiring malaria. Children who will be traveling to malaria-endemic regions should receive pretravel counseling on avoiding insects and appropriate chemoprophylaxis (AII) (526,527). Recommendations for chemoprophylaxis are the same for HIV-infected persons as for noninfected persons and are available at the CDC website (http://www.cdc.gov/malaria).
Potential exists for antiretroviral and antimalarial drug-drug interactions. Specifically, mefloquine significantly decreases steady-state ritonavir area-under-the-curve plasma levels by 31%. In children receiving a boosted PI regimen with preexisting moderate resistance, this decrease may be significant. In addition, antimalarial medications may need special preparation, and some are not easily delivered to children. Therefore, patients planning to travel to malaria-endemic areas are advised to visit a travel medicine specialist with training and experience in pediatrics at least 2 weeks before departure (AII).
Although TMP-SMX prophylaxis, in combination with use of bed nets, resulted in >90% reduction of clinical malaria in nonholoendmic areas in one study (528), CDC does not recommend TMP-SMX as an antimalarial prophylactic regimen. HIV-infected travelers must not rely on TMP-SMX for chemoprophylaxis against malaria because TMP--SMX has not been studied in areas of high malaria transmission, and TMP--SMX most likely will not provide adequate protection (DIII).
Discontinuing Primary Prophylaxis
Travel-related chemoprophylaxis with chloroquine, mefloquine, and doxycycline usually should be discontinued 4 weeks after departure from a malaria-endemic area because these drugs are not effective against malarial parasites developing in the liver and kill the parasite only once it has emerged to infect the red blood cells. Atovaquone-proguanil should be discontinued 1 week after departure from malaria-endemic areas. Chemoprophylaxis is not 100% effective, and malaria should be included in the differential diagnosis of fever or other signs or symptoms consistent with malaria in anyone who traveled to malaria-endemic areas during the previous 12 months.
Treatment Recommendations
Treatment of Disease
Treatment of malaria is based on the severity of disease, patient's age at onset, parasite species, and known resistance patterns (AI). HIV infection status does not affect choice of therapy (AII), and no recommendations exist for alternative dosing of antimalarial drugs in HIV-infected persons. Whether response to antimalarial treatment differs in regard to HIV infection status is unclear, but it does not appear to be appreciably different in young children (BIII). HIV-infected or HIV-exposed children frequently are excluded from drug efficacy trials because the children are receiving TMP--SMX prophylaxis. Published studies have reported conflicting results, have used older antimalarials, or were not adequately powered to answer this question (529--531). Treatment dosing for adolescents and children is provided in Table 4.
P. falciparum
Uncomplicated chloroquine-sensitive P. falciparum malaria should be treated with chloroquine. The recommended treatment options for uncomplicated chloroquine-resistant P. falciparum in the United States are atovaquone-proguanil (Malarone(r)), quinine with clindamycin or doxycycline (in children aged ≥8 years), or mefloquine (Lariam(r)) (AI). For dosages, refer to the CDC website above. It is imperative that the clinician choose a medication according to known sensitivity patterns from the area where the malaria was acquired.
The current drug of choice for uncomplicated chloroquine-resistant P. falciparum malaria is atovaquone-proguanil, which is FDA-approved for use in children weighing >5 kg, is well tolerated, has a wide therapeutic window, and provides simple dosing (pediatric tablets are available) (AIII). Although mefloquine is FDA-approved for persons aged >6 months, pediatric tablets are not available. However, mefloquine is the only treatment choice for uncomplicated chloroquine-resistant P. falciparum malaria in children weighing <5 kg.
Severe P. falciparum should be treated with IV quinidine gluconate (or IV quinine when available). Duration of quinine/quinidine therapy is typically 3 days with clindamycin, or doxycycline, continued for 7 days (AI). IV quinidine can cause hypoglycemia, ventricular arrhythmia, and QT prolongation; close monitoring, including telemetry, is required during infusion. Increasingly, hospital pharmacies do not always stock quinidine. Ritonavir inhibits quinidine metabolism and is therefore contraindicated in patients being treated with quinidine. An alternative is artesunate, which is available from CDC as an investigational new drug protocol (532). When signs or symptoms exist of severe disease, especially with indicators of a poor prognosis (including clinical features of impaired level of consciousness, respiratory distress, jaundice, seizures, or shock or laboratory features of hypoglycemia, elevated bilirubin, acidosis, elevated liver aminotransferase levels, or renal insufficiency), a tropical medicine specialist should be consulted. For artesunate release or additional assistance, contact the 24-hour CDC malaria hotline at 770-488-7788 during the day and 770-488-7100 after hours and on weekends and holidays.
P. vivax, P. ovale, P. malariae
The medication of choice for non-P. falciparum malaria is chloroquine; the parasite usually is sensitive to this drug (AI). Chloroquine usually is well tolerated. The most common reaction is self-limited itching (2%). However, among persons of African descent, the rate of this reaction is substantially higher (50%). In certain areas---most notably in New Guinea---P. vivax has known high rates of chloroquine resistance. A patient infected with known or suspected chloroquine-resistant P. vivax malaria should receive an alternative first-line agent (i.e., quinine plus clindamycin or doxycycline, atovaquone-proguanil, mefloquine). Both P. vivax and P. ovale have an intrahepatic stage (hypnozoite) that is not treated with the medications mentioned for the acute blood stage. To prevent relapse of P. vivax or P. ovale infection, patients should receive presumptive antirelapse therapy with primaquine after the primary treatment for the blood stage (AI). Glucose-6-phosphate dehydrogenase deficiency must be excluded before any use of primaquine because of the risk for severe hemolytic anemia.
Unknown species
When reliable identification of the malaria species is not possible or in a severely ill person, clinicians always should treat for the worst-case scenario of chloroquine-resistant P. falciparum malaria (AIII). PCR assays are now commercially available to speciate when microscopy is not sufficient. Assistance with speciation also is available from state public health departments and the CDC Division of Parasitic Diseases diagnostic service (http://www.dpd.cdc.gov/dpdx). After completion of initial therapy, knowing the malaria species is important because presumptive antirelapse therapy is necessary for P. ovale and P. vivax.
Monitoring and Adverse Events, Including IRIS
Severe malaria commonly induces hypoglycemia in children, especially when treated with IV quinine/quinidine because of the inhibition of gluconeogenesis and induction of endogenous insulin production. Therefore, use of a crystalloid solution containing glucose for fluid maintenance is prudent while closely monitoring glucose levels until IV quinine/quinidine therapy has been completed. Monitoring glucose is especially important for infants and for persons with altered mental status. Cardiac and intensive-care monitoring is recommended because quinine/quinidine can cause hypotension and may widen the QRS interval. Another common (50%--75%) adverse reaction to quinine is tinnitus, although this usually resolves after treatment. HIV-infected children may have more hematologic side effects, particularly neutropenia and anemia, following antimalarial therapy. IRIS caused by malaria has not been reported.
Management of Treatment Failure
Treatment failure with P. falciparum among children receiving a full course of appropriate antimalarial therapy occurs uncommonly. Patients should be monitored for clinical response and for signs of recrudescence after therapy completion. Published studies do not have the power to detect a difference in treatment outcomes between HIV-infected and HIV-uninfected children (529,530). Relapse of P. vivax and P. ovale can occur from the dormant (hypnozoite) liver form but is less common with primaquine treatment. Malaria medications purchased in sub-Saharan Africa or Southeast Asia may be counterfeit (533). When treatment failure occurs, malaria speciation should be confirmed, as should the geography of where the malaria was acquired. Retreatment with an appropriate first-line regimen should be given. Discussion with a tropical medicine expert or a call to the CDC malaria hotline is appropriate when complex situations arise.
Prevention of Recurrence
Except for reactivation of P. vivax and P. ovale hypnozoites, malaria, once successfully treated, does not recur. Malaria infection does not infer protective immunity and continued exposure to malaria parasites can result in repeated infection.
Discontinuing Secondary Prophylaxis
Not applicable.
Toxoplasmosis
Epidemiology
The major mode of transmission of Toxoplasma gondii infection among infants and young children is congenital, occurring almost exclusively among neonates born to women who sustain primary Toxoplasma infection during pregnancy. The estimated incidence of congenital toxoplasmosis in the United States is one case per 1,000--12,000 live-born infants (534,535). The seroprevalence of T. gondii in U.S.-born persons aged 12--49 years declined from 14.1% in the National Health and Nutrition Examination Survey 1988--1994 to 9.0% in 1999--2004 (536). Older children, adolescents, and adults typically acquire Toxoplasma infection by eating poorly cooked meat that contains parasitic cysts or by unintentionally ingesting sporulated oocysts in soil or contaminated food or water (537). Cats are the only definitive host for T. gondii. However, cats excrete sporulated oocysts in their feces only transiently after initial infection, and studies have failed to show a correlation between cat ownership and Toxoplasma infection in humans. Indeed, Toxoplasma infection in humans in the United States has declined despite increased cat ownership (537).
The overall risk for maternal-fetal transmission in HIV-uninfected women who acquire primary Toxoplasma infection during pregnancy is 29% (95% confidence interval [CI]: 25%--33%) (538). The risk for congenital infection is low among infants born to women who become infected during the first trimester (range: 2%--6%) but increases sharply thereafter, with a risk as high as 81% for women acquiring infection during the last few weeks of pregnancy (538,539). Infection of the fetus in early gestation usually results in more severe disease than does infection late in gestation.
The prevalence of latent Toxoplasma infection among women with and without HIV infection in the United States was assessed in a cross-sectional study of 2525 nonpregnant women enrolled in the Women's Interagency Health Study (540). The prevalence of Toxoplasma seropositivity was 15% and did not differ by HIV infection status. A few cases of mother-to-infant transmission of Toxoplasma in HIV-infected women have been reported (541--545). Prenatal transmission of T. gondii from women without HIV infection who have chronic Toxoplasma infection acquired before pregnancy is rare (546). However, with HIV coinfection, perinatal transmission of Toxoplasma has been observed among women with chronic Toxoplasma infection (transmission rate: <4%), presumably because of reactivation of replication of the organism among women with severe immune suppression (541--544).
CNS infection with T. gondii was reported as an AIDS-indicator condition in <1% of pediatric AIDS cases before the advent of HAART (547). During the HAART era, this condition is rarely encountered in U.S. children with HIV infection. Development of CNS toxoplasmosis in HIV-infected children during the HAART era occurred in 0.2% (five of 2767) of those enrolled in the long-term follow-up study PACTG 219c (3). In most cases of Toxoplasma encephalitis (TE) among HIV-infected children, infection is considered to have occurred in utero. More rarely, it has been reported among older HIV-infected children, who presumably had primary acquired toxoplasmosis (548--550). As in adults, the greatest risk is among severely immunosuppressed children (e.g., CD4 count <50 cells/mm3).
Clinical Manifestations
In studies of nonimmunocompromised infants with congenital toxoplasmosis, most infants (70%--90%) are asymptomatic at birth. However, most asymptomatic children develop late sequelae (e.g., retinitis, visual impairment, and intellectual or neurologic impairment), with onset of symptoms ranging from several months to years after birth. Symptoms in newborns take either of two presentations: generalized disease or predominantly neurologic disease. Symptoms might include maculopapular rash; generalized lymphadenopathy; hepatosplenomegaly; jaundice; hematologic abnormalities, including anemia, thrombocytopenia, and neutropenia; and substantial CNS disease, including hydrocephalus, intracerebral calcification, microcephaly, chorioretinitis, and seizures (551).
Similarly, toxoplasmosis acquired after birth is most often initially asymptomatic. When symptoms occur, they are frequently nonspecific and can include malaise, fever, sore throat, myalgia, lymphadenopathy (cervical), and a mononucleosis-like syndrome featuring a maculopapular rash and hepatosplenomegaly.
TE should be considered among all HIV-infected children with new neurologic findings. Although focal findings are more typical, the initial presentation can vary and reflect diffuse CNS disease. Other symptoms include fever, reduced alertness, and seizures.
Isolated ocular toxoplasmosis is rare and usually occurs in association with CNS infection. As a result, a neurologic examination is indicated for children in whom Toxoplasma chorioretinitis is diagnosed. Ocular toxoplasmosis appears as white retinal lesions with little associated hemorrhage; visual loss might occur initially.
Less frequent presentations among HIV-infected children with reactivated chronic toxoplasmosis include systemic toxoplasmosis, pneumonitis, hepatitis, and cardiomyopathy/myocarditis (544,552).
Diagnosis
HIV-infected women might be at increased risk for transmitting T. gondii to their fetuses, and serologic testing for Toxoplasma should be performed for all HIV-infected pregnant women. All infants whose mothers are both HIV-infected and seropositive for Toxoplasma should be evaluated for congenital toxoplasmosis (90). Congenital toxoplasmosis can be diagnosed by EIA or an immunosorbent assay to detect Toxoplasma-specific IgM, IgA, or IgE in neonatal serum within the first 6 months of life or persistence of specific IgG antibody beyond age 12 months (553--556). IgA might be more sensitive for detecting congenital infection than IgM or IgE (554). However, approximately 20%--30% of infants with congenital toxoplasmosis will not be identified during the neonatal period with IgA or IgM assays (555).
Serologic testing is the major method of diagnosis, but interpretation of assays often is confusing and difficult. Using the services of a specialized reference laboratory that can perform serology, isolation of organisms, and PCR and offers assistance in interpreting results, especially when attempting to diagnose congenital toxoplasmosis, can be helpful (554).
Additional methods that can be used to diagnose infection in the newborn include isolation of the Toxoplasma parasite by mouse inoculation or inoculation in tissue cultures of CSF, urine, placental tissue, amniotic fluid, or infant blood. T. gondii DNA can be detected by PCR performed on clinical specimens (e.g., white blood cells, CSF, amniotic fluid, or tissue) in a reference laboratory (554,555). If a possible diagnosis of congenital toxoplasmosis at delivery is uncertain, the neonate should be evaluated, including ophthalmologic, auditory, and neurologic examinations; lumbar puncture; and imaging of the head (either CT or magnetic resonance imaging [MRI] scans) to determine whether hydrocephalus or calcifications are present.
In the United States, routine Toxoplasma serologic screening of HIV-infected children whose mothers do not have toxoplasmosis is not recommended because of its low prevalence. However, in regions with high incidence of Toxoplasma infection (≥1% per year), serologic testing might be selectively considered for HIV-infected children aged >12 months. HIV-infected adolescents without previous Toxoplasma infection should undergo serologic testing.
CNS toxoplasmosis is presumptively diagnosed on the basis of clinical symptoms, serologic evidence of infection, and presence of a space-occupying lesion on imaging studies of the brain (557). TE rarely has been reported in persons without Toxoplasma-specific IgG antibodies; therefore, negative serology does not definitively exclude that diagnosis. CT of the brain might indicate multiple, bilateral, ring-enhancing lesions in CNS toxoplasmosis, especially in the basal ganglia and cerebral corticomedullary junction. MRI is more sensitive and will confirm basal ganglia lesions in most patients (558). F-fluoro-2-deoxyglucose--positive emission tomography reportedly is helpful in adults in distinguishing Toxoplasma abscesses from primary CNS lymphoma, but the accuracy is not high, and this test is not widely available.
Definitive diagnosis of TE requires histologic or cytologic confirmation by brain biopsy, which might demonstrate leptomeningeal inflammation, microglial nodules, gliosis, and Toxoplasma cysts. Brain biopsy is reserved by some experts for patients who do not respond to specific therapy.
Prevention Recommendations
Preventing Exposure
All HIV-infected children and adolescents and their caregivers should be counseled about sources of T. gondii infection. They should be advised not to eat raw or undercooked meat, including undercooked lamb, beef, pork, or venison (BIII). All meat (lamb, beef, pork, and chicken) should be cooked to an internal temperature of 165°F--170°F (559) until it is no longer pink inside. Hands should be washed after contact with raw meat and after gardening or other contact with soil; in addition, fruits and vegetables should be washed well before being eaten raw (BIII). Stray cats should not be handled or adopted; a cat already in the household should be kept inside and the litter box changed daily, preferably by an HIV-negative, nonpregnant person (BIII). Cats should be fed only canned or dried commercial food or well-cooked table food, not raw or undercooked meats (BIII). Patients need not be advised to part with their cats or to have their cats tested for toxoplasmosis (EII).
Preventing First Episode of Disease
Toxoplasma-seropositive adolescents and adults who have a CD4 count of <100 cells/mm3 should be administered prophylaxis against TE (AII) (435). Specific levels of immunosuppression that increase the risk for TE in children are less well defined. Toxoplasma-seropositive children with CD4 <15% should be administered prophylaxis against TE (AIII). For children aged ≥6 years, the same absolute CD4 cell count level used for HIV-infected adults can be used (AIII).
In HIV-infected adolescents and adults, the double-strength tablet daily dose of TMP-SMX recommended as the preferred regimen for PCP prophylaxis is effective TE prophylaxis (AII) (435). TMP--SMX, one double-strength tablet three times weekly (or 3 consecutive days a week), is an alternative (BIII). Data are limited on the efficacy of TMP--SMX as a primary preventive agent for TE in children. However, data on adults suggest it also is the regimen of choice in children (BIII) (Table 1). If patients cannot tolerate TMP--SMX, the recommended alternative is dapsone-pyrimethamine, which also is effective against PCP (BI) (560,561). Atovaquone with or without pyrimethamine also can be considered (CIII). Single-drug prophylaxis with dapsone, pyrimethamine, azithromycin, or clarithromycin cannot be recommended (DII). Aerosolized pentamidine does not protect against TE and is not recommended (EI) (435,436). Toxoplasma-seronegative adults and adolescents who are not taking a PCP-prophylactic regimen known to be active against TE should be retested for IgG antibody to Toxoplasma when their CD4 count declines to <100 cells/mm3 to determine whether they have seroconverted and are therefore at risk for TE (CIII). Severely immunosuppressed children who are not receiving TMP--SMX or atovaquone who are seropositive for Toxoplasma should be administered prophylaxis for both PCP and toxoplasmosis (i.e., dapsone plus pyrimethamine) (BIII).
Discontinuing Primary Prophylaxis
Prophylaxis against TE should be discontinued among HIV-infected adults and adolescents who have responded to HAART with an increase in CD4 count to >200 cells/mm3 for >3 months (AI). Multiple observational studies (447,562,563) and two randomized trials (564,565) have reported that primary prophylaxis can be discontinued with minimal risk for TE recrudescence among patients who have responded to HAART with an increase in CD4 count from <200 cells/mm3 to ≥200 cells/mm3 for >3 months. Although patients with CD4 counts of <100 cells/mm3 are at greatest risk for TE, the risk for TE when CD4 count increases to 100--200 cells/mm3 has not been studied as rigorously as an increase to >200 cells/mm3. Thus, the recommendation specifies discontinuing prophylaxis after an increase to >200 cells/mm3. Discontinuing primary TE prophylaxis is recommended because prophylaxis adds limited disease prevention for toxoplasmosis and because discontinuing drugs reduces pill burden, the potential for drug toxicity, drug interactions, selection of drug-resistant pathogens, and cost. Data do not exist on the safety of discontinuing primary TE prophylaxis for HIV-infected children whose CD4 percentage rises above 15%. Data on adults suggest discontinuation of TMP-SMX may be safe once a child responds to HAART with a sustained rise in CD4 percentage above 15%; for children aged ≥6 years, the same CD4 count used for HIV-infected adults can be used (CIII).
Prophylaxis should be reintroduced in HIV-infected adults and adolescents if the CD4 count decreases to <100--200 cells/mm3 (AIII) or the CD4 percentage falls below 15% for HIV-infected children (BIII).
Treatment Recommendations
Treatment of Disease
Pregnant women with suspected or confirmed primary toxoplasmosis and newborns with possible or documented congenital toxoplasmosis should be managed in consultation with an appropriate specialist. Although controversy exists about the efficacy of treating pregnant women who have acute toxoplasmosis in an attempt to prevent infection of the fetus (566), most experts would recommend such therapy (BII) (90). For an HIV-infected woman with a symptomatic Toxoplasma infection during pregnancy, empiric therapy of the newborn should be strongly considered, regardless of whether the mother was treated during pregnancy (BIII).
The preferred treatment for congenital toxoplasmosis is pyrimethamine combined with sulfadiazine, with supplementary leucovorin (folinic acid) to minimize pyrimethamine-associated hematologic toxicity (AII) (551,567). Although the optimal duration of therapy is undefined, the recommended duration of treatment of congenital toxoplasmosis for infants without HIV infection is 12 months (AII) (567).
For HIV-uninfected children who have mild congenital toxoplasmosis, certain experts alternate pyrimethamine/sulfadiazine/folinic acid monthly with spiramycin during months 7--12 of treatment (CIII). However, for children with moderate to severe disease and children with HIV infection, the full 12-month regimen of pyrimethamine/sulfadiazine should be administered (AII).
HIV-infected children with acquired CNS, ocular, or systemic toxoplasmosis should be treated with pyrimethamine and leucovorin plus sulfadiazine (AI). Acute therapy should be continued for 6 weeks, assuming clinical and radiologic improvement (BII). Longer courses of treatment might be required for extensive disease or poor response after 6 weeks. The primary alternative for sulfadiazine in patients who develop sulfonamide hypersensitivity is clindamycin, administered with pyrimethamine and leucovorin (AI). Azithromycin instead of clindamycin also has been used with pyrimethamine and leucovorin among sulfa-allergic adults, but this regimen has not been studied among children (CIII).
Another alternative in adults is atovaquone plus pyrimethamine and leucovorin, or atovaquone with sulfadiazine alone, or atovaquone as a single agent among patients intolerant to both pyrimethamine and sulfadiazine (BII); however, these regimens have not been studied in children (CIII). TMP-SMX alone has been used as an alternative to pyrimethamine-sulfadiazine in adults but not in children (CIII).
For isolated ocular toxoplasmosis in nonimmunocompromised hosts, TMP-SMX alone is as effective as pyrimethamine-sulfadiazine (568). However, these data have not been duplicated in HIV-infected persons; therefore, this regimen cannot be recommended for this group of patients (DIII).
Corticosteroids (e.g., dexamethasone or prednisone) are recommended for children with CNS disease when CSF protein is highly elevated (i.e., >1000 mg/dL) or with focal lesions with substantial mass effects (BIII). Because of the potential immunosuppressive effects of steroids, they should be discontinued as soon as possible.
Anticonvulsants should be administered to children with TE who have a history of seizures (AIII) but should not be administered prophylactically to children without a history of seizures (DIII). Anticonvulsants, if administered, should be continued at least through acute therapy.
Monitoring and Adverse Events, Including IRIS
Children with TE should be routinely monitored for clinical and radiologic improvement and for adverse effects of treatment; changes in antibody titers are not useful for monitoring responses to therapy.
Toxoplasmosis-associated IRIS has been described rarely in HIV-infected adults and has not been described in HIV-infected children (569).
Pyrimethamine can be associated with rash (including Stevens-Johnson syndrome) and nausea. The primary toxicity of pyrimethamine is reversible bone marrow suppression (i.e., neutropenia, anemia, and thrombocytopenia). A complete blood count should be performed at least weekly while the child is on daily pyrimethamine and at least monthly while on less-than-daily dosing (AIII). Leucovorin (folinic acid) always should be administered with pyrimethamine; increased doses of leucovorin might be required in the event of marrow suppression. Because of the long half-life of pyrimethamine, leucovorin should be continued 1 week after pyrimethamine has been discontinued.
Adverse effects of sulfadiazine include rash, fever, leukopenia, hepatitis, GI symptoms (e.g., nausea, vomiting, and diarrhea), and crystalluria. Clindamycin can be associated with fever, rash, and GI symptoms (e.g., nausea; vomiting; and diarrhea, including pseudomembranous colitis) and hepatotoxicity.
Drug interactions between anticonvulsants and antiretrovirals should be evaluated. Patients receiving corticosteroids should be closely monitored for development of other OIs.
Management of Treatment Failure
Brain biopsy should be considered in the event of early clinical or radiologic neurologic deterioration despite adequate empiric treatment or in children who do not clinically respond to anti-Toxoplasma therapy after 10--14 days. For children who undergo brain biopsy and have confirmed histopathologic evidence of TE despite treatment, a switch to an alternative regimen as previously described should be considered (BIII).
Prevention of Recurrence
Patients who have completed initial therapy for acquired TE should be administered lifelong suppressive therapy (i.e., secondary prophylaxis or chronic maintenance therapy) (AI) (570,571) unless immune reconstitution occurs with antiretroviral therapy (Table 3). The combination of pyrimethamine plus sulfadiazine plus leucovorin is highly effective for this purpose (AI). A commonly used regimen for patients who cannot tolerate sulfa drugs is pyrimethamine plus clindamycin (BI); however, only the combination of pyrimethamine plus sulfadiazine provides protection against PCP as well (AII). Data on adults indicate atovaquone with or without pyrimethamine also can be considered for children (CIII). Limited data support the use of TMP-SMX for secondary prophylaxis (572); this regimen should be used only for persons who do not tolerate pyrimethamine plus sulfadiazine or pyrimethamine plus clindamycin (CII).
Discontinuing Secondary Prophylaxis
Adults and adolescents receiving secondary prophylaxis for acquired TE are at low risk for recurrence of TE when they have successfully completed their initial therapy, continue to have no signs or symptoms of TE, and have a sustained increase in CD4 count of >200 cells/mm3 after HAART (e.g., >6 months) (562,563,565,573,574). Discontinuing chronic maintenance therapy in HIV-infected adolescents and adults who meet these criteria is a reasonable consideration (BI). Highest risk for relapse appears to occur within the first 6 months after stopping secondary prophylaxis. Certain specialists would obtain an MRI of the brain as part of their evaluation to determine whether discontinuing therapy is appropriate. The safety of discontinuing secondary prophylaxis after immune reconstitution with HAART among children has not been studied extensively. However, given the data in adults, clinicians caring for HIV-infected children aged 1--5 years can consider discontinuing secondary prophylaxis against T. gondii after they have completed TE therapy and ≥6 months of stable HAART and are asymptomatic and once the CD4 percentage has risen to ≥15% for >3 consecutive months. For children aged ≥6 years, the same CD4 count used in adults (CD4 >200 cells/mm3) also can be used (BIII). Prophylaxis should be reinstituted if these parameters are not met.
Viral Infections
Cytomegalovirus
Epidemiology
Infection with human cytomegalovirus (CMV) is common and usually inapparent; CMV can be acquired during infancy, early childhood, or adolescence. Transmission can occur vertically from an infected woman to her offspring or horizontally by contact with virus-containing breast milk, saliva, urine, or sexual fluid or through transfusion of infected blood or transplantation of infected organs. During infancy and early childhood, infection usually occurs secondary to ingestion of infected breast milk or exposure to infected saliva or urine. Infection occurs at younger ages in locations where sanitation is less than optimal. Among adolescents, sexual transmission is the major mode of CMV acquisition.
Age-related prevalence of infection varies widely, depending on living circumstances and social customs. Breast-feeding, child-rearing practices, crowding, sanitation, and sexual behavior most likely influence age-related variations in CMV prevalence. Where rates of maternal seropositivity are high and breast-feeding is common, more than half of infants acquire CMV during the first year of life (575). Group care of children facilitates spread of CMV, especially in toddlers, and leads to higher prevalence of infection in children who attend child care centers and in their caregivers (576,577). In Africa, Asia, and Latin America, most children are infected by CMV before adolescence. In the United States and western Europe, the prevalence of antibody to CMV in adults from middle and upper socioeconomic strata is 40%--60%, whereas the prevalence in low-income adults is ≥80% (578). Overall, among U.S. women of childbearing age, the prevalence of CMV infection is 50%--80%, with the highest prevalence in women in lower socioeconomic strata (579,580). The prevalence of CMV infection among HIV-infected pregnant women is higher than in the general population, with approximately 90% of HIV-infected pregnant women coinfected with CMV (581,582).
CMV is the most common congenitally transmitted infection, occurring in 0.2%--2.2% of live-born infants in the United States (583). Congenital (in utero) CMV infection occurs most commonly among infants born to women who have primary CMV infection during pregnancy. Following primary infection during pregnancy, the rate of transmission to the fetus is approximately 30%--40% (579,584). In comparison, the rate of congenital infection after nonprimary CMV infection usually is believed to be significantly lower (range: 0.15%--1.0%) (583,585,586). More recent studies demonstrate that in utero transmission of nonprimary maternal infection can occur because of reactivation of infection among women infected before pregnancy or reinfection with a different CMV strain among CMV-seropositive women (587,588); these studies challenge the traditional understanding of transmission risk and clinical outcomes following nonprimary maternal CMV infection.
CMV also can be transmitted during the intrapartum or postpartum periods from mother to infant. Up to 57% of infants whose mothers shed CMV at or around delivery become infected with CMV, and up to 53% of children who are breast-fed milk containing infectious virus can become CMV infected. Symptomatic CMV disease in the infant is much less common when CMV is acquired intrapartum or through breast-feeding than when acquired antenatally and occurs primarily in premature neonates.
HIV-infected women with CMV infection have a higher rate of CMV shedding from the cervix than do women without HIV infection (52%--59% and 14%--35%, respectively) (589). The risk for mother-to-infant transmission of CMV may be higher among infants born to women dually infected with CMV and HIV. In one study of 440 infants born to HIV-infected U.S. women, the overall rate of in utero infection was 4.5% (11), higher than the <2% rate of in utero infection in the general U.S. population.
HIV-infected children appear to be at higher risk for CMV infection during early childhood than do HIV-uninfected children (11). The rate of CMV acquisition in HIV-infected children appears to be particularly high during the first 12 months of life but remains higher among HIV-infected than HIV-uninfected children through age 4 years when they are exposed to CMV infection in other children in child care or school.
CMV disease occurs less frequently among HIV-infected children than HIV-infected adults but contributes substantially to morbidity and mortality. In the pre-antiretroviral era, CMV caused 8%--10% of pediatric AIDS-defining illness (590). Data in HIV-infected adults have shown a 75%--80% decrease in the incidence of new cases of CMV end-organ disease with the advent of antiretroviral therapy, with an incidence now estimated to be <6 cases per 100 person-years (591). In a study of OIs in approximately 3000 children followed in PACTG studies during the pre-HAART era, the frequency of CMV retinitis was 0.5 cases per 100 child-years and, of other CMV disease, 0.2 cases per 100 child-years (1). The rate varied significantly by CD4 percentage; the incidence of CMV retinitis was 1.1 cases per 100 child-years in children with CD4 <15%, compared with 0.1 case per 100 child-years in children with CD4 >25%. In data from the same cohort during the HAART era, the overall rate of CMV retinitis was <0.5 per 100 child-years (3). In the Perinatal AIDS Collaborative Transmission Study, the incidence of nonocular CMV before and after January 1997 (pre- and post-HAART eras) was 1.4 per 100 child-years and 0.1 per 100 child-years, respectively (4). Similarly, CMV retinitis declined from 0.7 to 0.0 per 100 child-years during the same period.
Symptomatic HIV-infected children coinfected with CMV have a higher rate of CMV viruria than do asymptomatic HIV-infected or HIV-exposed children. Overall, up to 60% of children with AIDS shed CMV. This compares with one third of HIV-infected children shedding CMV; 15%--20% of CMV-infected, HIV-exposed but uninfected children; and <15% of CMV-infected infants not exposed to HIV (592).
Clinical Manifestations
Approximately 10% of infants with in utero CMV infection are symptomatic at birth with congenital CMV syndrome (CMV inclusion disease); mortality of children with symptomatic disease is as high as 30%. About half of neonates with symptomatic congenital CMV disease are small for gestational age; additional findings may include petechiae (76%), jaundice (67%), hepatosplenomegaly (60%), chorioretinitis (20%), microcephaly (53%), intracranial calcifications (55%), and hearing impairment (≤65%) (593,594). Approximately 90% of infants with symptomatic disease at birth who survive have late complications, including substantial hearing loss, mental retardation, chorioretinitis, optic atrophy, seizures, or learning disabilities (579). Although most children with in utero CMV infection do not have symptoms at birth, 10%--15% are at risk for later developmental abnormalities, sensorineural hearing loss, chorioretinitis, or neurologic defects.
HIV-infected children coinfected with CMV appear to have faster progression of HIV disease than do those without CMV infection (11,590,595). In one study from the pre-HAART era, 53% of infants coinfected with HIV and CMV had progression to AIDS or had died by age 18 months, compared with 22% of HIV-infected children without CMV infection; those with HIV/CMV coinfection also were more likely to have CNS manifestations (36% versus 9%). The relative risk for HIV disease progression in children coinfected with CMV compared with children without CMV was 2.6 (95% CI: 1.1--6.0) (11).
CMV retinitis is the most frequent severe manifestation of CMV disease among HIV-infected children, accounting for approximately 25% of CMV AIDS-defining illnesses. CMV retinitis among young HIV-infected children is frequently asymptomatic and discovered on routine examination. Older children with CMV retinitis present similarly to adults, with floaters, loss of peripheral vision, or reduction in central vision. Diagnosis of CMV retinitis is based on clinical appearance with white and yellow retinal infiltrates and associated retinal hemorrhages. A more indolent, granular retinitis also can occur. HIV-infected children with CD4 counts <100 cells/mm3 are more likely than those with higher CD4 counts to develop CMV retinitis; however, CD4 count is less predictive of risk for CMV disease in young infants, and systemic and localized CMV disease can occur in HIV-infected infants with higher, age-adjusted CD4 counts (592,596).
End-organ CMV disease has been reported in the lung, liver, GI tract, pancreas, kidney, sinuses, and CNS (596--599). In children with extraocular CMV disease, predominantly nonspecific symptoms (e.g., fever, poor weight gain, and loss of developmental milestones with laboratory abnormalities of anemia, thrombocytopenia, and elevated lactic dehydrogenase) are initially observed, although the extent to which CMV or HIV infection themselves contribute to these findings is unclear (592). GI manifestations among HIV-infected children include CMV colitis (the most common GI manifestation), oral and esophageal ulcers, hepatic involvement, ascending cholangiopathy, or gastritis. Odynophagia is a common presentation of CMV esophagitis, whereas abdominal pain and hematochezia frequently occur with CMV colitis. Sigmoidoscopy in CMV colitis is nonspecific, demonstrating diffuse erythema, submucosal hemorrhage, and diffuse mucosal ulcerations. Esophageal or colonic ulcerations may cause perforation or hemorrhage.
The role of CMV in pulmonary disease among HIV-infected children is difficult to assess because it often is isolated with other organisms (e.g., P. jirovecii). Histologic evidence of CMV disease is needed to determine whether active disease is present. CMV pneumonia is an interstitial process with gradual onset of shortness of breath and dry, nonproductive cough; auscultatory findings may be minimal.
CNS manifestations of CMV include subacute encephalopathy, myelitis, and polyradiculopathy (primarily observed in adults but rarely reported in children). The subacute or chronic encephalopathy of CMV can be difficult to differentiate clinically from HIV dementia, with symptoms of confusion and disorientation attributable to cortical involvement. Focal signs can be attributed to lesions in the brainstem. CSF findings are nonspecific and might indicate a polymorphonuclear predominance (>50% of patients), elevated protein (75%), and low glucose (30%). However, up to 20% of children with CMV CNS involvement have completely normal CSF indices. CMV also can cause a rapidly progressive, often fatal, CNS disease with defects in cranial nerves, nystagmus, and increasing ventricular size (600).
Diagnosis
CMV infection (versus disease) can be difficult to differentiate in HIV-infected children. Because of transplacental transfer of antibody from mother to child, a positive CMV antibody assay in an infant aged <12 months can indicate maternal infection but not necessarily infection of the infant. In an infant aged >12 months, a positive CMV antibody assay indicates previous infection with CMV but not necessarily active disease. In children of any age, a positive CMV culture or PCR indicates infection but not necessarily disease.
CMV can be isolated in cell culture from peripheral blood leukocytes, body fluids (e.g., urine), or tissues. Using centrifugation-assisted shell vial culture amplification techniques, CMV can be detected within 16--40 hours of culture inoculation. A positive blood buffy-coat culture establishes CMV infection and increases the likelihood that disease or symptoms were caused by CMV because children with positive blood cultures are at higher risk for end-organ disease.
Different methods have been used to detect viral antigen or DNA directly and to identify patients at risk for CMV disease, including detection of pp65 antigenemia, qualitative and quantitative PCR, and DNA hybridization. The DNA assays are more sensitive than buffy-coat or urine cultures for detecting CMV and can be used to identify patients at higher risk for clinically recognizable disease. CMV DNA detection in CSF by DNA PCR is highly sensitive for CMV CNS disease. Quantitative DNA PCR can be used as a marker of risk for disease and to monitor response to therapy (601).
Recovery of virus from tissues (e.g., endoscopically guided biopsies of GI or pulmonary tissue) provides evidence of disease in symptomatic patients. The limitation of this method is that detection of visible cytopathic effects in cell culture takes 1--6 weeks. Staining of shell vial culture with CMV monoclonal antibodies or immunostaining for CMV antigens can allow earlier diagnosis of infection. Histopathology demonstrates characteristic "owl's eye" intranuclear and smaller intracytoplasmic inclusion bodies in biopsy specimens; staining with CMV monoclonal antibodies or immunostaining for CMV antigens also can be done. The same procedures can be used on cells obtained from bronchoalveolar lavage.
Prevention Recommendations
Preventing Exposure
HIV-exposed infants and HIV-infected children, adolescents, and adults who are seronegative for CMV and require blood transfusion should be administered only CMV antibody-negative or leukocyte-reduced cellular blood products in nonemergency situations (BIII).
Annual CMV antibody testing is recommended beginning at 1 year of age for CMV-seronegative HIV-infected infants and children who are severely immunosuppressed (e.g., CD4 count <100 cells/mm3 or CD4 percentage <10%) (BII). Annual testing allows identification of children who have acquired CMV infection and might benefit from screening for retinitis.
HIV-infected adults and adolescents who are child care providers or parents of children in child care facilities should be informed that they are at increased risk for CMV infection (BI). Risk for CMV infection can be diminished by optimal hygienic practices (e.g., hand-washing) (AII).
Preventing First Episode of Disease
The primary method for preventing severe CMV disease is recognition of the early manifestations of the disease and prevention of severe immunosuppression by treating with HAART. HIV-infected children aged <5 years who are CMV-infected and severely immunosuppressed (e.g., CD4 count <50 cells/mm3 or CD4 percentage <5%) should have a dilated retinal examination performed by an ophthalmologist every 6 months (AIII). Older children should be counseled to be aware of floaters in the eye and visual changes, similar to the recommendation for adults (BIII). During the HAART era, CMV end-organ disease has diminished to such an extent that primary prophylaxis with antiviral agents in CMV and HIV coinfected people usually is not recommended (DIII). CMV end-organ disease is best prevented by antiretroviral therapy to maintain CD4 count >100 cells/mm3. If this is not possible, prophylaxis with valganciclovir can be considered for HIV-infected adolescents who are CMV-seropositive, have a CD4 count of <50 cells/mm3, and weigh enough to receive adult doses of valganciclovir (CIII).
Discontinuing Primary Prophylaxis
Because primary prophylaxis with antiviral agents in persons coinfected with CMV and HIV is not recommended (as discussed above), no consideration of discontinuing primary prophylaxis is necessary.
Treatment Recommendations
Treatment of Disease
Treatment of newborns who have symptomatic congenital CMV disease involving the CNS with IV ganciclovir for 6 weeks has been evaluated in a series of clinical trials conducted by the National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group (602,603); all infants in these studies were HIV-uninfected. Infants receiving therapy cleared their urine of CMV by culture by the end of the 6-week treatment period, although all experienced a rebound in their viruria after antiviral discontinuation (602). In a phase III, randomized, controlled trial, infants receiving IV ganciclovir for 6 weeks were less likely to have hearing deterioration over the first 2 years of life than were infants receiving no antiviral therapy (603). Treated infants also had more rapid resolution of liver enzyme abnormalities and a greater degree of growth during the course of therapy. They also experienced fewer neurodevelopmental delays at 1 year of life than did nontreated infants (604). However, approximately two thirds of the infants developed substantial neutropenia during therapy (603). Among patients developing neutropenia, 48% required dose modification, but most were able to complete the 6 weeks of therapy. On the basis of these results, IV ganciclovir therapy (6 mg/kg/dose administered every 12 hours) for 6 weeks should be offered to HIV-exposed or HIV-infected infants who have symptomatic congenital CMV disease involving the CNS (BI). If during the 6 weeks of therapy an infant is confirmed as HIV-infected, some experts would recommend longer duration of treatment (>6 weeks) (BIII).
CMV retinitis should be managed in concert with an experienced ophthalmologist. IV ganciclovir, oral valganciclovir, IV foscarnet, IV cidofovir, and the ganciclovir intraocular implant coupled with valganciclovir are all effective treatments for CMV retinitis in HIV-infected adults (AI) (605--609). For HIV-infected children, the drug of choice for initial treatment for CMV retinitis---and for other end-organ disseminated CMV disease (e.g., colitis, esophagitis, and CNS disease)---is IV ganciclovir (AI). Oral valganciclovir, a prodrug of ganciclovir, is one of the first-line treatments for HIV-infected adults with CMV retinitis (AI) (607). The drug is well absorbed from the GI tract and rapidly metabolized to ganciclovir in the intestine and liver. However, data on appropriate dosage of this drug for children are limited (610). Additionally, a valganciclovir liquid formulation is not commercially available. Even though extemporaneously compounded valganciclovir "recipes" are available, the pharmacokinetics, bioavailability, safety, and shelf-life of such formulations are unknown, and they should not be used in children. Thus, oral valganciclovir is an option primarily for older children who weigh enough to receive the adult dose and tablet formulation of valganciclovir (CIII).
An alternative drug for treating CMV disease or for use in ganciclovir-resistant CMV infections in HIV-infected children is foscarnet (AI). Foscarnet used as suppressive therapy has been associated with increased length of survival relative to ganciclovir in HIV-infected adults. Doses should be modified among patients with renal insufficiency.
Combination therapy with ganciclovir and foscarnet delays progression of retinitis in certain patients in whom monotherapy fails (596,607,611,612) and can be used as initial therapy among children with sight-threatening disease (BIII). Combination therapy also has been used for adults with retinitis that has relapsed on single-agent therapy. Combination therapy with IV ganciclovir and foscarnet also can be considered in initial therapy of CMV CNS disease (BII). However, substantial rates of adverse effects are associated with combination therapy.
Before the availability of valganciclovir, oral ganciclovir in combination with an intraocular ganciclovir implant had been used for maintenance treatment of CMV retinitis in adults. Given the lack of commercial availability of oral ganciclovir, its use in children can no longer even be considered.
In adults, the combination of oral valganciclovir with a ganciclovir sustained-release intraocular implant, replaced every 6--9 months, was superior to daily IV ganciclovir in preventing relapse of retinitis and is preferred by some adult HIV specialists for patients who have CMV lesions adjacent to the optic nerve or fovea (AI) (605--609). This regimen can be considered for treatment and chronic suppression of CMV retinitis in older children who weigh enough to receive the adult dose and tablet formulation of valganciclovir.
Cidofovir is effective in treating CMV retinitis among adults who are intolerant of other therapies. However, cidofovir has not been studied in children with CMV disease (CIII).
Intravitreous injections of ganciclovir, foscarnet, or cidofovir have been used to control retinitis but require biweekly intraocular injections. Data are limited in children, and biweekly injection is impractical for use in most children (DIII). Implantation of an intravitreous ganciclovir medication-release device in the posterior chamber of the eye also has been used in HIV-infected adults and adolescents. In HIV-infected adults with CMV retinitis, ganciclovir intraocular implant plus oral valganciclovir is superior to once daily IV ganciclovir for preventing relapse of CMV retinitis (605--607). Intraocular implant plus IV ganciclovir or oral valganciclovir may be the preferred initial treatment for patients with immediate sight-threatening infections (e.g., adjacent to the optic nerve or fovea). Small peripheral lesions can be treated with systemic therapy without local treatment (BII). Intraocular implants should not be used in children aged <3 years because of the small size of their eyes (EIII). Intraocular cidofovir is not recommended in children because of lack of data and the risk for hypotonia in adults (EIII).
For CMV neurologic disease, prompt initiation of therapy is critical for an optimal clinical response. However, levels of ganciclovir in the CNS are 24%--70% of plasma levels, and levels in the brain are approximately 38% of plasma levels (613); hence, combination treatment with ganciclovir and foscarnet might be preferred as initial therapy to stabilize disease and maximize response (BII). However, this approach is associated with substantial rates of adverse effects, and optimal treatment for neurologic disease in children receiving optimized HAART is unknown.
Monitoring and Adverse Events, Including IRIS
CMV retinitis should be managed in concert with an experienced ophthalmologist. Recommendations for HIV-infected adults include indirect ophthalmoscopy through a dilated pupil performed at diagnosis of CMV retinitis, after completion of induction therapy, 1 month after initiation of therapy, and monthly thereafter while the patient is on anti-CMV treatment; recommendations should be similar for HIV-infected children with CMV retinitis (AIII). Monthly fundus photographs, using a standardized photographic technique that documents the appearance of the retina, provide the optimum method for following patients and detecting early relapse (AIII). For patients who have experienced immune recovery (Table 3), the frequency of ophthalmologic follow-up can be decreased to every 3 months. However, because relapse of the retinitis occurs among patients with immune recovery, regular ophthalmologic follow-up still is needed.
The major side effects of ganciclovir and valganciclovir are myelosuppression (i.e., anemia, neutropenia, and thrombocytopenia) and renal toxicity. Dose reduction or interruption because of hematologic toxicity might be necessary in up to 40% of patients receiving IV ganciclovir; granulocyte colony-stimulating factor can be used to ameliorate marrow suppression. The main toxicities of foscarnet are decreased renal function and metabolic derangements. Renal toxicity and foscarnet binding to divalent metal ions, such as calcium, lead to metabolic abnormalities in approximately one third of patients, and serious electrolyte imbalances (including abnormalities in calcium, phosphorus, magnesium, and potassium levels) and secondary seizures, cardiac dysrhythmias, abnormal liver transaminases, and CNS symptoms can occur. Metabolic disturbances can be minimized if foscarnet is administered by slow infusion, with rates not exceeding 1 mg/kg/minute. Concomitant use of other nephrotoxic drugs increases the likelihood of renal dysfunction associated with foscarnet therapy. For patients receiving ganciclovir or foscarnet, complete blood counts and serum electrolytes and renal function should be monitored twice weekly during induction therapy and once weekly thereafter (AIII).
The major side effect of cidofovir is potentially irreversible nephrotoxicity; the drug produces proximal tubular dysfunction including Fanconi syndrome and acute renal failure. Renal toxicity manifests as proteinuria and glycosuria. To minimize nephrotoxicity, probenicid should be administered before each infusion, and IV hydration with normal saline should be administered before and after each cidofovir infusion. For patients receiving IV cidofovir, blood urea nitrogen, creatinine, and urinalysis should be performed before each infusion; administration of the drug is contraindicated if renal dysfunction or proteinuria is detected. Other reported adverse events include anterior uveitis and ocular hypotony; serial ophthalmologic monitoring for anterior segment inflammation and intraocular pressure is needed while receiving the drug systemically. Cidofovir should not be administered concomitantly with other nephrotoxic agents. Cidofovir therapy must be discontinued if serum creatinine increases ≥0.5 mg/dL above baseline.
Immune recovery uveitis after initiation of effective HAART is an immunologic reaction to CMV associated with inflammation in the anterior chamber and/or the vitreous (614). Ocular complications of uveitis include macular edema and development of epiretinal membranes, which can cause loss of vision. Immune recovery uveitis may respond to periocular corticosteroids or a short course of systemic steroids. Oral valganciclovir was beneficial in one small uncontrolled study (615).
Management of Treatment Failure
Resistant strains of CMV should be suspected when progressive disease and continued recovery of virus occurs despite ganciclovir therapy. Foscarnet is the drug of choice when ganciclovir resistance is suspected (AI).
In patients with CMV retinitis, although drug resistance occurs among patients receiving long-term therapy, early relapse might be caused by the limited intraocular penetration of systemically administered drugs; in HIV-infected adults, the placement of a ganciclovir implant in a patient whose retinitis has relapsed during systemic treatment is recommended because it achieves greater drug levels in the eye and often will control the retinitis for 6--8 months until the implant requires replacement (BIII). Because of the size requirements of the implants, this option would be limited to older children with CMV retinitis. Many experts would initially treat early first relapse of retinitis with reinduction with the same drug, followed by reinstitution of maintenance therapy (AII). However, if drug resistance is suspected or if side effects or toxicities interfere with optimal courses of the initial agent, change to an alternative drug is reasonable (AIII). Combination ganciclovir and foscarnet can be considered but is accompanied by greater toxicity.
Prevention of Recurrence
Courses of antiviral agents (e.g., ganciclovir, foscarnet, or cidofovir) do not cure CMV disease. After induction therapy, the standard recommendation has been to provide secondary prophylaxis (chronic maintenance therapy) for the remainder of the patient's life (AI). Regimens that can be considered for chronic suppression in adults and adolescents include IV ganciclovir, oral valganciclovir, IV foscarnet, combined IV ganciclovir and foscarnet, parenteral cidofovir, and (for retinitis only) ganciclovir administration by intraocular implant (AI) (616--623). Because of limited data on drug pharmacokinetics and dosing in children, IV ganciclovir or foscarnet are the preferred secondary prophylaxis regimens for children; oral valganciclovir can be considered for older children able to receive adult dosing. Repetitive intravitreous injections of ganciclovir, foscarnet, and cidofovir reportedly are effective for secondary prophylaxis of CMV retinitis (624,625), although intraocular therapy alone does not protect the contralateral eye or other organ systems and therefore typically is combined with systemic treatment (616). Additionally, frequent intravitreous injections are impractical for use in most children (DIII).
A chronic maintenance regimen for patients treated for CMV disease should be chosen in consultation with a specialist. Chronic maintenance therapy is not routinely recommended for GI disease but should be considered if relapses occur (BII). A role for maintenance therapy for CMV pneumonitis has not been established (CIII). For patients with retinitis, decisions should be made in consultation with an ophthalmologist and should take into consideration the anatomic location of the retinal lesion, vision in the contralateral eye, and the immunologic and virologic status of the patient (BIII). Intraocular implants should not be used in children aged <3 years because of the small size of their eyes (EIII).
Discontinuing Secondary Prophylaxis
Multiple case series have reported that maintenance therapy can be discontinued safely in adults and adolescents with CMV retinitis whose CD4 counts have increased substantially in response to HAART (626--631). These patients have remained disease free for >30 to 95 weeks, whereas during the pre-HAART era, retinitis typically reactivated in <6 to 8 weeks after stopping CMV therapy. Plasma HIV RNA levels varied among these patients, supporting the hypothesis that the CD4 count is the primary determinant of immune recovery to CMV. CMV retinitis can occur in HAART-treated adults with high CD4 counts, however (632), suggesting that CMV-specific cellular immunity may be important in controlling CMV in immune-reconstituted HIV-infected adults (633,634). In HIV-infected adults with CMV retinitis, discontinuing secondary prophylaxis is considered for patients with a sustained increase in CD4 count to >100 cells/mm3 in response to treatment.
The safety of discontinuing secondary prophylaxis after immune reconstitution with HAART in HIV-infected children has not been as well studied. Low or undetectable HIV replication in children is the strongest correlate with CMV immune reconstitution, being associated with a higher frequency of CMV-specific CD4 cells (635). Early institution of HAART may help control CMV infection by maintaining normal CD4 count and cytotoxic T-lymphocyte responses in HIV-infected children (636). In deciding whether to discontinue secondary prophylaxis, the significant toxicities associated with antiviral drugs active against CMV, including those in in vitro and animal models, need to be considered.
Recognizing the limitations of the data in children but drawing on the growing experience in adults, discontinuing prophylaxis may be considered for children aged 1--5 years who are receiving HAART and have a sustained (e.g., >6 months) increase in CD4 count to >500 cells/mm3 or CD4 percentage to >15%, and for children aged ≥6 years, an increase in CD4 count to >100 cells/mm3 as for adults (CIII). Such decisions should be made in close consultation with an ophthalmologist and should account for such factors as magnitude and duration of CD4 cell increase, anatomic location of the retinal lesion, vision in the contralateral eye, and the feasibility of regular ophthalmologic monitoring (CIII).
All patients in whom anti-CMV maintenance therapy has been discontinued should continue to undergo regular ophthalmologic monitoring at at least 3- to 6-month intervals for early detection of CMV relapse and for immune reconstitution uveitis (AII). CMV viral load or other markers of CMV infection (e.g., antigenemia or viral DNA tests) are not well standardized; their role in predicting relapse remains to be defined, and they are not recommended for routine monitoring (DIII) (637,638).
Reinitiating Secondary Prophylaxis
CMV retinitis relapses among adults whose anti-CMV maintenance therapies have been discontinued and whose CD4 counts have decreased to <50 cells/mm3 (624); reinstitution of secondary prophylaxis is recommended for HIV-infected adults when CD4 count falls to <100 cells/mm3. For HIV-infected children in whom secondary prophylaxis has been discontinued because of immune reconstitution, secondary prophylaxis should be reinstituted in children aged 1--5 years when the CD4 count decreases to <500 cells/mm3 or CD4 percentage to <15%, and for children aged ≥6 years when the CD4 count decreases to <100 cells/mm3 or CD4 percentage to <15% (BIII).
Hepatitis B Virus
Epidemiology
Chronic hepatitis B infection is defined as persistence of hepatitis B surface antigen (HbsAg) for >6 months. Risk of developing chronic hepatitis B infection after acute infection correlates with age at time of hepatitis B virus (HBV) infection. Among HBV-infected persons, chronic hepatitis B infection develops in ≤90% of infants, 25%--50% of children aged 1--5 years, and 6%--10% of older children and adolescents (639--641).
Childhood HBV infection can be acquired parenterally, perinatally, or through sexual transmission. Horizontal transmission of HBV can occur secondary to frequent interpersonal contact of nonintact skin or mucus membranes with blood or body fluids that contain blood (e.g., saliva) or from sharing household objects, such as toothbrushes or razors. Universal hepatitis B vaccination of newborns has dramatically lowered chronic HBV infection among children and reduced the rates of HBV-related morbidity and mortality in the United States.
Adolescents are at risk for HBV infection through sexual activity or injection-drug use. In a study of HIV-infected adolescents at 43 PACTG centers, 19% had evidence of current or resolved HBV infection; the rate of current or resolved HBV infection in HIV-infected adolescent girls was twice the U.S. population-based rates for HIV-uninfected adolescent girls and, for adolescent boys, nearly seven times higher (642). Substance abuse and sexual activity increase the risk for HIV/HBV coinfection in adolescents, particularly among males who have sex with other males (643).
Most children who acquire HBV perinatally are immunotolerant to HBV. Although they have high HBV DNA levels, serum transaminase levels are usually normal, and necroinflammatory liver disease is minimal. Childhood-acquired HBV infection is characterized by lower HBV DNA levels, greater serum transaminase elevation, and higher necroinflammatory liver disease than in perinatally acquired HBV infection (644).
Data from the National Health and Nutrition Examination Survey, 1999--2004, indicate that 0.51% (95% CI: 0.3%--0.9%) of children aged 6--19 years had ever been infected with HBV (645). Data are limited on the prevalence of chronic HBV infection in HIV-infected children in the United States. In a study of 228 HIV-infected children at an inner-city hospital, six HIV-infected children had chronic HBV infection (2.6% [95% CI: 1.1%--5.9%]) (646). The mean age of HIV/HBV-coinfected children was 17 years; 33% had acquired both HIV and HBV infection through blood transfusion; and 84% were HBV early antigen-positive (HBeAg), indicating infectiousness. Half of coinfected children had normal serum transaminase levels, and half had mild elevations of up to twofold above upper limit of normal.
Clinical Manifestations
Most acute HBV infections in children are asymptomatic. Prodromal symptoms of lethargy, malaise, fatigue, nausea, and anorexia can occur. Jaundice and right upper quadrant pain can follow and, less commonly, hepatomegaly and splenomegaly. Gianotti-Crosti syndrome (papular acrodermatitis), urticaria, macular rash, or purpuric lesions may be seen in acute HBV infection. Extrahepatic manifestations associated with circulating immune complexes that have been reported in HBV-infected children include arthralgias, arthritis, polyarteritis nodosa, thrombocytopenia, and glomerulonephritis. Most children with chronic HBV infection are asymptomatic. However, rare cases of fulminant hepatic failure have occurred during childhood HBV infection (647).
Most children with chronic HBV infection are asymptomatic. One quarter of infants and children with chronic HBV eventually will develop cirrhosis or hepatocellular carcinoma (HCC) (648,649). However, these sequelae usually develop over 2--3 decades and rarely occur during childhood (650). Development of HCC correlates with HBV DNA levels and duration of HBV infection, with the highest risk in persons infected in early life (651). HIV/HBV-coinfected adults are at increased risk for cirrhosis, end-stage liver disease, and liver-related mortality (652).
Diagnosis
Testing for HBV infection should be performed in any child whose mother is known to be infected with HBV. Adolescents and young adults with histories of injection-drug use or high-risk sexual contact also should undergo testing for HBV infection.
HBsAg is the first marker detectable in serum, appearing 30 days after infection; it precedes the elevation of serum aminotransferase levels and the onset of symptoms. Necroinflammatory liver disease then can occur, during which serum transaminase levels increase, along with high HBV DNA levels and HBeAg positivity. HBeAg correlates with viral replication, DNA polymerase activity, infectivity, and increased severity of liver disease. Antibody to hepatitis B core antigen (anti-HBc) appears 2 weeks after HBsAg and persists for life. Passively transferred maternal anti-HBc can be detectable in the infant up to age 12 months. In self-limited infections, HBsAg is usually eliminated in 1--2 months, and hepatitis B surface antibody (anti-HBs) develops during convalescence. Anti-HBs indicates immunity from HBV infection. After recovery from natural infection, both anti-HBs and anti-HBc usually are present. In persons who become chronically infected (i.e., persistently positive for HBsAg beyond 24 weeks), anti-HBs is not detectable. Persons who have been vaccinated may have detectable anti-HBs but not anti-HBc.
HBeAg seroconversion, defined as loss of HBeAg, followed by the production of antibodies to HBeAg (e.g., anti-HBe), heralds transition of the HBV-infected person to the inactive carrier state. HBeAg seroconversion is infrequent in children aged <3 years and may not occur until the third or fourth decade of HBV infection (653). Less than 10% of children infected perinatally with HBV undergo HBeAg seroconversion. In contrast, higher rates of HBeAg seroconversion occur in childhood-acquired HBV infection, with 70%--80% of children acquiring anti-HBe by the second decade of life (650). HBeAg seroconversion usually is followed by reduction of serum HBV DNA levels, an initial increase and then subsequent normalization of serum transaminase levels, followed by resolution of necroinflammatory liver disease (650). Development of cirrhosis and HCC occur more commonly in patients with delayed HBeAg seroconversion (654). HBeAg-negative infection (precore mutant) occurs uncommonly in children (641).
HBV DNA is a marker for HBV replication. In the active phase of chronic hepatitis B, high HBV DNA levels have been associated with necroinflammatory liver disease. Children infected perinatally, however, may remain in an immunotolerant phase with high levels of HBV DNA without evidence of liver damage. Quantitative DNA assays may help determine the need for treatment and for evaluating treatment response. Although not necessary for diagnostic purposes, liver biopsy may be useful to assess the degree of liver damage and determine the need for treatment.
Prevention Recommendations
Prevention of Exposure
All pregnant women should be tested for HBsAg during an early prenatal visit in each pregnancy (AI). Testing should be repeated in late pregnancy for HBsAg-negative women at high risk for HBV infection (e.g., injection-drug users, women with intercurrent sexually transmitted infections, and women with multiple sex partners). Pregnancy is not a contraindication to hepatitis B vaccination for women who have not previously been vaccinated; current hepatitis B vaccines contain noninfectious HBsAg and should cause no risk to the fetus.
Preventing First Episode of Disease
All infants born to HBV-infected women, including HIV-coinfected women, should receive hepatitis B vaccine and hepatitis B immune globulin (HBIG) within 12 hours after birth, a second dose of hepatitis B vaccine at age 1--2 months, and a third dose at age 6 months (A1) (Figures 1 and 2) (38). For preterm infants weighing <2000 g, the initial vaccine dose (birth dose) should not be counted as part of the vaccine series because of the potentially reduced immunogenicity of hepatitis B vaccine in these infants; three additional doses of vaccine (for a total of four doses) should be administered beginning when the infant reaches age 1 month (AI) (38). A three-dose hepatitis B vaccine regimen is 95% effective in preventing HBV infection in HBV-exposed infants. Postvaccination testing for anti-HBs and HBsAg should be performed at age 9--18 months in infants born to HBsAg positive women. The level of anti-HBs that is considered protective is >10 mIU/mL. Infants who are HBsAg negative and have anti-HBs levels <10 mIU/mL should be revaccinated with a second three-dose series of hepatitis B vaccine and retested 1--2 months after the final vaccine dose (38).
The three-dose series of hepatitis B vaccine also is recommended for all children and adolescents aged <19 years who were not previously vaccinated. However, antibody responses to hepatitis B vaccination may be diminished in HIV-infected children, especially in older children or children with CD4 counts <200 cells/mm3 (655,656). For this reason, HIV-infected infants, children, and adolescents should be tested for anti-HBs 1--2 months after completing the vaccination series and, if anti-HBs levels are <10 mIU/mL, revaccinated with a second three-dose series of hepatitis B vaccine (AIII). Modified hepatitis B vaccine dosing regimens, including a doubling of the standard antigen dose, might increase response rates. However, although a current randomized trial is evaluating use of various hepatitis B vaccine preparations and doses in HIV-infected youth, no data are available yet.
The need for booster doses of hepatitis B vaccine in HIV-infected persons has not been determined. Annual anti-HBs testing and booster doses when the anti-HBs levels decline to <10 mIU/mL should be considered in persons with ongoing risk for hepatitis B exposure (CIII) (38).
In addition to hepatitis B vaccine, hepatitis A vaccine can prevent hepatitis infection and its potential devastating long-term sequelae and thus all children should receive hepatitis A vaccination at age 12--23 months with the two doses in the series administered ≥6 months apart (39). Children who are not fully vaccinated by age 2 years can be vaccinated at subsequent visits. Hepatitis A vaccine is also recommended for children aged ≥24 months who live in areas where vaccination programs target older children or those who are at increased risk for infection (Figures 1 and 2).
HBV-infected children should be advised not to share toothbrushes or other personal-care articles that might be contaminated with blood. Although efficiency of sexual transmission of HBV is relatively low, safe-sex practices should be encouraged for all HIV-infected adolescents and young adults; barrier precautions (e.g., latex condoms) are recommended to reduce the risk for exposure to sexually transmitted pathogens, including HBV.
Treatment Recommendations
Treatment of Disease
General issues
Individualization of therapy is essential for any HBV-infected child and should be based on the child's age, age at acquisition of infection, HBV DNA levels, and serum transaminase levels. Antiviral therapy regimens for chronic hepatitis B are approved only for children aged >2 years who have compensated liver disease.
HIV-infected children who are not receiving anti-HBV therapy should be closely monitored with determination of serum aminotransferase levels every 6 months. If serum transaminase levels are persistently elevated (more than twofold the upper limit of normal for ≥6 months), HBeAg, anti-HBe, and HBV DNA levels should be obtained. Monitoring of serum transaminases and HBV DNA levels over time is important before the initiation of antiviral therapy to identify patients who may be in the process of spontaneous HBeAg seroconversion who would not require treatment. Liver biopsy is not required before treatment but may help to determine the severity of hepatic inflammation and fibrosis and to exclude other causes of liver disease.
No clear recommendations exist for treating chronic childhood HBV infection. HBV-infected children often have milder disease than adults and may show spontaneous HBeAg seroconversion. Few large randomized controlled trials exist of antiviral therapies for chronic hepatitis B infection in childhood. Moreover, the long-term safety of many of the agents used to treat chronic hepatitis B infection in adults is not known in children. However, a 2004 consensus meeting of pediatric liver experts recommended that antiviral treatment be considered in children with chronic HBV infection and a duration of necroinflammatory liver disease >6 months (657).
Indications for treatment of chronic HBV infection in HIV-coinfected children are the same as in HBV-infected children without HIV infection: 1) evidence of ongoing HBV viral replication, as indicated by detectable serum HBV DNA, with or without HBeAg positivity, for >6 months and persistent elevation of serum transaminase levels (at least twice the upper limit of normal for >6 months); or 2) evidence of chronic hepatitis on liver biopsy (BII) (641,657). Children without necroinflammatory liver disease usually do not warrant antiviral therapy (DIII). Treatment is not recommended for children with immunotolerant chronic HBV infection (i.e., normal serum transaminase levels despite detectable HBV DNA) (DIII). The goals of treatment for children with chronic hepatitis B infection are identical to those for adults: suppression of HBV replication; normalization of serum transaminase levels; acceleration of HBeAg seroconversion; preservation of liver architecture; and prevention of long-term sequelae, such as cirrhosis and HCC.
The optimal agent and duration of therapy for childhood hepatitis B infection remain unclear. Treatment of chronic hepatitis B infection is evolving; consultation with providers with expertise in treating chronic hepatitis B infection in children is recommended.
Treatment of chronic hepatitis B infection in adults and adolescents
Seven medications have been approved to treat chronic hepatitis B infection in adults: interferons (both standard and pegylated); nucleoside analogues (i.e., lamivudine; telbivudine; and entecavir); and the nucleotide analogues, adefovir and tenofovir. The FDA-approved HIV antiretroviral medication emtricitabine also has significant activity against HBV, although it is not approved for this indication. Preferred initial therapies for adults who have chronic hepatitis B without HIV infection include pegylated interferon-alfa, entecavir, or adefovir monotherapy. In adults who have chronic hepatitis B infection with or without HIV infection, treatment for hepatitis B is considered in HBeAg-positive persons with HBV DNA ≥20,000 IU/mL (>105 copies/mL), HBeAg-negative persons with HBV DNA ≥2000 IU/mL (>104 copies/mL), patients with persistent serum transaminase elevation, or patients with evidence of cirrhosis or fibrosis (652).
Treatment options for HBV in HIV-infected persons must account for the goals of therapy and the impact treatment might have on both HIV and HBV replication. In coinfected patients who require treatment for chronic hepatitis B, HIV, or both, many experts would initiate a fully suppressive regimen to treat HIV infection that includes a dual nucleoside analogue backbone with drugs active against both HIV and HBV plus a third agent active against HIV; this approach might reduce the risk for IRIS, particularly in patients with advanced immune deficiency. Tenofovir plus lamivudine or emtricitabine would be the first choice for the nucleoside backbone; the combination of tenofovir with lamivudine was demonstrated to be more effective in suppressing HBV than either drug alone and prevents development of lamivudine resistance (658). In instances in which HIV treatment is not an option but treatment of hepatitis B infection is needed, pegylated interferon-alfa can be used alone because it does not lead to development of drug-resistant HIV or HBV mutants. Use of tenofovir, lamivudine, or emtricitabine without a fully suppressive HAART regimen should be avoided because of the rapid development of drug-resistant HIV mutations.
Treatment of chronic hepatitis B infection in children without HIV infection
Only two drugs (monotherapy with interferon-alfa [standard] or lamivudine) are FDA-approved to treat chronic hepatitis B in children (AI). The limited pediatric trials of these agents show that although they are well-tolerated by children, response rates are low, and treatment does not fully eliminate HBV infection (659,660). In HIV-uninfected children, HBeAg seroconversion rates after 1 year of treatment are similar (641). Interferon-alfa treatment is administered for only 6 months but requires subcutaneous administration and has more frequent side effects, including growth impairment. Although lamivudine is administered orally and has a lower rate of side effects, it requires a longer duration of therapy and has a high rate of resistance if taken for an extended time (641).
Although various combination regimens involving sequential or concurrent lamivudine and standard or pegylated interferon-alfa have been studied in children or adults with chronic hepatitis B, the superiority of combination therapy over monotherapy with standard or pegylated interferon-alfa or lamivudine has not been demonstrated; however, lamivudine resistance rates may be lower (661--670). A recent study of children with immunotolerant HBV infection suggested possible benefit from sequential lamivudine and interferon-alfa therapy, with 78% of patients clearing HBV DNA by the end of treatment (664). However, combination therapy cannot be recommended for pediatric HBV infection until more data are available (DII).
Treatment of HBV/HIV-coinfected children
None of the clinical studies of treatment of chronic hepatitis B infection have specifically studied children with HIV/HBV coinfection. As in coinfected adults, choice of antiviral therapy for the HIV/HBV-coinfected child involves consideration of whether concurrent HIV treatment is warranted.
- If treatment of chronic hepatitis B but not HIV infection is indicated, standard interferon-alfa is the preferred agent (BIII). Adefovir also could be considered in older children able to receive adult dosing (BIII). Antiviral drugs with activity against HIV (e.g., lamivudine, emtricitabine, tenofovir, and possibly entecavir) should be avoided to prevent future development of drug-resistant HIV mutations.
- If treatment of HIV infection but not chronic hepatitis B is indicated, use of a HAART regimen that avoids drugs with activity against HBV (e.g., lamivudine, emtricitabine, or tenofovir) is recommended to prevent future development of HBV drug resistance (BIII). Alternatively, in older coinfected children who can receive tenofovir, use of a HAART regimen with a nucleoside analogue backbone that contains two drugs effective against HBV (tenofovir plus lamivudine or emtricitabine) can be considered (BIII).
- If treatment for both HIV and chronic hepatitis B is indicated and the child is lamivudine-naïve, an antiretroviral regimen that includes lamivudine (or emtricitabine) is recommended (BIII). A regimen containing tenofovir and a nucleoside analogue (either lamivudine or emtricitabine) is preferred for HIV/HBV-coinfected adults, and should be considered for use in older HIV-infected children or adolescents who can receive adult dosage. However, tenofovir is not approved for use in HIV-infected children <18 years, and no pediatric formulations are available. Although pediatric studies with an investigational pediatric formulation of tenofovir are under way, data are not yet available.
- If treatment for HIV and chronic hepatitis B is indicated and the child is receiving antiretroviral therapy including lamivudine or emtricitabine with HIV suppression but detectable plasma HBV DNA, HBV lamivudine resistance can be assumed. However, HBV drug-resistant isolates may have lower replicative capacity, and some experts recommend continued use of lamivudine or emtricitabine, although this recommendation is controversial (CIII). Treatment options for such children who require HBV therapy include the addition of interferon therapy to the antiretroviral regimen (BIII), or tenofovir (BIII), or adefovir if the child can receive adult dosing (BIII). Data are insufficient on other anti-HBV drugs in children to make recommendations.
Interferons
Standard interferon-alfa-2a or -2b has received the most study in children who have chronic hepatitis B (without HIV infection) and is recommended for treating chronic hepatitis B infection with compensated liver disease in HIV-uninfected children aged ≥2 years who warrant treatment (BII). In a review of six randomized clinical trials in 240 HBV-infected children aged >1.5 years, interferon-alfa therapy resulted in HBV DNA clearance in 35% of treated children, HBeAg clearance in 10%, and normalization of serum transaminase levels in 39% at treatment completion (671). Six to 18 months after therapy discontinuation, 29% of children had persistent HBV DNA, and 23% demonstrated HBeAg clearance. Children most likely to respond to interferon treatment are of younger age, higher baseline serum transaminase levels, and lower baseline HBV DNA levels (659,672--674). Response is less likely (10%) in those with normal serum transaminase levels, high HBV DNA levels, HBV genotypes C or D, or HBeAg-negative chronic HBV infection. Interferon-alfa therapy might be considered to treat chronic hepatitis B in HIV-coinfected children who do not require antiretroviral therapy for their HIV infection (BIII).
The standard course of interferon-alfa therapy for children without HIV infection is 24 weeks. Pegylated interferon-alfa, which results in more sustained plasma interferon concentrations and can be administered by injection once weekly for 48 weeks, has proven superior to standard interferon-alfa in treating HBV-infected adults (667,675). However, the limited data on use of pegylated interferon-alfa in children come from treatment of hepatitis C infection, and appropriate dosing information is not available for use of pegylated interferon-alfa to treat chronic hepatitis B in children (DIII) (676--678).
Lamivudine
Lamivudine (3TC) is an oral nucleoside analogue that inhibits HBV replication. It is approved for use in children aged 2--17 years who have compensated liver disease from chronic hepatitis B. In a placebo-controlled trial in children who have chronic hepatitis B without HIV infection, lamivudine was well tolerated, with virologic response (clearance of HBV DNA and HBeAg) in 23% of children receiving 52 weeks of lamivudine therapy, compared with 13% in placebo recipients (679). Response rates were higher (35%) for children with baseline serum transaminases more than two times normal (679). In a 2-year, open-label extension of this study, 213 children who remained HBeAg-positive after 1 year of therapy were continued on lamivudine treatment; virologic response was seen in 21% of the original lamivudine recipients, compared with 30% of prior placebo recipients, indicating that additional clinical response could occur over time with prolonged treatment (680). However, longer duration of lamivudine therapy also was associated with progressive development of lamivudine-resistant HBV, with base pair substitutions at the tyrosine-methionine-aspartate-aspartate (YMDD) locus of HBV DNA polymerase.
Accordingly, lamivudine should not be used as a single agent for treatment of chronic hepatitis B in HIV-infected children because of the risk for HIV resistance to lamivudine (EIII); as discussed above, lamivudine should be used only in HIV/HBV-coinfected children in combination with other antiretroviral drugs in a HAART regimen (BIII). The dose of lamivudine required to treat HIV infection is higher than to treat pediatric chronic hepatitis B alone; therefore, the higher dose of lamivudine should be used in HIV/HBV-coinfected children to avoid development of lamivudine-resistant HIV (AIII). Lamivudine resistance should be suspected if HBV DNA levels increase during antiviral therapy. Such increases may precede increases in serum transaminase levels (hepatic flare) and liver decompensation (674).
Emtricitabine
Emtricitabine is structurally similar to lamivudine and is active against HBV and HIV, although not approved for treatment of chronic hepatitis B. Like lamivudine, emtricitabine also is associated with relatively rapid onset of HBV and HIV drug resistance, and patients with suspected lamivudine resistance should be assumed to have cross-resistance to emtricitabine. Lamivudine and emtricitabine should be considered interchangeable for treatment of chronic hepatitis B and not additive (BIII). As with lamivudine, emtricitabine should not be used to treat chronic hepatitis B in coinfected children who are not being treated with combination antiretroviral therapy for their HIV infection because of the risk for HIV-associated resistance mutations (EIII).
Adefovir
Adefovir dipivoxil is an oral nucleotide analogue active against HBV. Although active against HBV, adefovir has minimal anti-HIV activity, and HIV resistance has not been observed in patients receiving a 10-mg daily dose of adefovir for 48 weeks (681). HBV resistance to adefovir is much lower than in lamivudine, being reported as 2% after 2 years, 4% after 3 years, and 18% after 4 years of therapy in adults (682). These adefovir-associated mutations in HBV Pol gene result in only a modest (threefold to eightfold) increase in the 50% inhibitory concentration and are partially cross-resistant with tenofovir. Adefovir is now FDA-approved for adults who require treatment for chronic hepatitis B but do not yet require treatment for their HIV infection. Adefovir has been studied in HIV/HBV-coinfected adults with lamivudine-resistant HBV infection, and HBV suppression was demonstrated (681). Safety and effectiveness of adefovir for treating chronic hepatitis B in children has not yet been established, but an ongoing randomized clinical trial is evaluating its use in HIV-uninfected children aged 2--17 years who have chronic hepatitis B (659).
Tenofovir
Tenofovir is a nucleotide analog structurally similar to adefovir that reduces HBV DNA levels in adults with lamivudine-resistant and wild-type HBV infection. A study in HIV/HBV-coinfected adults receiving stable antiretroviral therapy comparing treatment with tenofovir or adefovir found similar efficacy in suppression of HBV DNA without differences in toxicity (683). Another study of HIV/HBV-coinfected adults receiving tenofovir in addition to lamivudine as part of their antiretroviral regimen found that HBV DNA became undetectable in 30% of HBeAg-positive and 82% of HBeAg-negative patients, most of whom had lamivudine-resistant HBV infection (681). As noted earlier, tenofovir is not approved for use in HIV-infected children aged <18 years, and no pediatric formulation is available. However, for HIV/HBV-coinfected adolescents who require treatment of both infections and who can receive adult doses, tenofovir in combination with an anti-HBV nucleoside (either lamivudine or emtricitabine) can be considered for treatment (BIII); a combined formulation of emtricitabine and tenofovir (Truvada) is available for adults. As with lamivudine and emtricitabine, tenofovir should not be used for treatment of chronic hepatitis B in HIV-coinfected patients who are not receiving combination antiretroviral therapy for their HIV infection because of the risk for HIV-associated resistance mutations (EIII).
Entecavir
Entecavir is an oral nucleoside analogue that inhibits HBV DNA polymerase. Compared with lamivudine, entecavir therapy results in greater HBV viral suppression, increased normalization of serum transaminase levels, improved liver histology, and lower HBV resistance rates (684). HBV viral suppression also has been demonstrated in HIV/HBV-coinfected adults. Entecavir treatment is approved for treatment of chronic hepatitis B in adults and is preferred for lamivudine-resistant HBV infections. However, it recently was demonstrated to have suppressive activity against HIV (685). Entecavir should not be used in HIV/HBV-coinfected patients who are not receiving combination antiretroviral therapy for their HIV infection. No data are available on safety and efficacy of entecavir in children.
Telbivudine
Telbivudine is a thymidine nucleoside analogue that was approved to treat chronic hepatitis B in adults. It is well tolerated, but like lamivudine, resistance emerges over time, and telbivudine is not active against lamivudine-resistant HBV. No data are available on telbivudine in HIV/HBV-coinfected adults. No data on children exist on safety and efficacy of telbivudine.
Duration of therapy
The optimal duration of therapy in HIV/HBV-coinfected children is not known. The duration of interferon-alfa treatment in HIV-uninfected children with chronic hepatitis B is 6 months. At least 1 year of lamivudine therapy is recommended for HIV-uninfected children who have chronic hepatitis B, with continuation of medication for ≥6 months after documented HBeAg seroconversion (659). However, because lamivudine would be administered only to HIV/HBV-coinfected children who need HIV treatment and as part of a suppressive antiretroviral regimen, treatment with lamivudine (or other anti-HBV drugs with anti-HIV activity) should be continued indefinitely in children with HIV/HBV coinfection, even in the instance of HBeAg seroconversion (CIII).
Monitoring and Adverse Events, Including IRIS
The parameters of successful therapy for chronic hepatitis B are not well defined, but markers of improvement include decreased hepatic necroinflammatory disease, normalization of serum transaminase levels, reduction of HBV DNA levels, and HBeAg seroconversion. In children starting treatment for chronic hepatitis B, serum transaminase levels should be measured every 3--6 months. If the child also is beginning HAART, some experts would monitor transaminase levels more frequently during the first few months of therapy (e.g., monthly for 3 months) because of the risk for IRIS (see below). Monitoring of response to treatment for chronic hepatitis B is based on testing for HBV DNA and HBeAg and anti-HBe antibody on the same schedule as transaminase evaluations (every 3--6 months). Among HBeAg-positive persons, treatment for chronic hepatitis B should be continued until HBeAg seroconversion has been achieved and ≥6 months of additional treatment has been completed after the appearance of anti-HBe (BIII). Close monitoring for relapse is needed after withdrawal of therapy. Among persons who are HBeAg negative, treatment should be continued until HBsAg clearance has been achieved.
In HIV/HBV-coinfected persons starting HAART, serum transaminase elevations ("flares") can occur as part of IRIS or secondary to HAART-associated hepatotoxicity. HBV-associated liver injury is thought to be immune-mediated, and restoration of immunocompetence with antiretroviral treatment may reactivate liver inflammation and damage. Initiation of HAART without anti-HBV therapy can lead to reactivation of HBV. This does not represent a failure of HAART but rather a sign of immune reconstitution. IRIS manifests by an increase in serum transaminase levels as the CD4 count increases during the first 6--12 weeks of HAART. Thus, serum transaminase levels should be monitored closely after introduction of HAART. In such situtations, HAART should be continued and treatment for HBV initiated. The prognosis for most IRIS cases is favorable because a robust inflammatory response may predict an excellent response to HAART in terms of immune reconstitution and, perhaps, improved survival. In a patient experiencing a hepatic flare, differentiating between IRIS and drug-induced liver toxicity may be difficult, and no reliable clinical or laboratory predictor exists to distinguish between the two. Close collaboration of the HIV specialist with a specialist in hepatic disease is recommended for such patients; a hepatologist should be consulted promptly if elevated aminotransferases levels are associated with clinical jaundice or other evidence of liver dysfunction (e.g., low serum albumin).
Clinical and laboratory exacerbations of hepatitis and hepatic flare can occur in children receiving HAART if agents with anti-HBV activity are discontinued. Some experts recommend that once antiretroviral drugs with anti-HBV activity are begun, they should be continued unless contraindicated or until the child has been treated for >6 months after HBeAg seroconversion and can be closely monitored after discontinuation (BIII). If discontinuation of therapy for chronic hepatitis B results in hepatic flare, therapy for chronic hepatitis B should be reinstituted (BIII).
Some clinicians recommend monitoring HBV-infected children or adolescents for HCC with baseline screening and then yearly determinations of serum alpha fetoprotein levels and abdominal ultrasonography; however, no data support the benefit of such surveillance (641,657,659,660).
Adverse effects of interferon-alfa use in children, although frequent, usually are not severe or permanent; however, approximately 5% of children require treatment discontinuation. The most common side effects include an influenza-like syndrome, cytopenias, and neuropsychiatric effects. Influenza-like symptoms comprising fever, chills, headache, myalgia, arthralgia, abdominal pain, nausea, and vomiting are seen in 80% of patients during the first month of treatment. These side effects decrease substantially during the first 4 months of therapy; premedication with acetaminophen or ibuprofen might reduce side effects. Subtle personality changes, which resolve when therapy is discontinued, have been reported in 42% of children (686). Depression and suicidal ideation have also been reported in clinical trials of children treated with interferon-alfa (687). Neutropenia, which resolves after discontinuation of therapy, is the most common laboratory abnormality; anemia and thrombocytopenia are less common. Abnormalities in thyroid function (hypothyroidism or hyperthyroidism) have been reported with interferon-alfa therapy (688). Loss of appetite with transient weight loss and impaired height growth might occur but usually resolves after completion of therapy (689). Less commonly observed side effects of interferon-alfa include epistaxis and transient mild alopecia. Antinuclear autoantibodies have been detected in some children treated with interferon-alfa. Interferon-alfa therapy is contraindicated for children with decompensated liver disease; severe cytopenia; severe renal, cardiac, or neuropsychiatric disorders; and autoimmune disease (EII) (690). Elevation of serum transaminase levels has been reported during interferon-alfa therapy in children and adults but usually is not an indication to stop therapy; these flares may herald impending HBeAg seroconversion (659). Children receiving interferon-alfa therapy should be monitored with a complete blood count and serum level of thyroid stimulating hormone at baseline and periodically (e.g., at least every 3 months) for the duration of treatment.
Lamivudine usually is well-tolerated in children; rare cases of lactic acidosis and pancreatitis have been reported in HIV/HBV-coinfected adults. Tenofovir and adefovir can cause renal tubular disease. Patients receiving either drug should have baseline urinalysis and periodic serum creatinine and phosphate monitoring. Administration of other nephrotoxic agents increases the risk for renal toxicity.
Management of Treatment Failure
Treatment failure is defined as ongoing HBV replication, persistent serum transaminase elevations, and the failure of HBeAg seroconversion in HBeAg-positive persons. Flares of liver disease with increasing HBV DNA levels can be seen with the development of resistance to lamivudine or emtricitabine.
In some children who have received initial treatment for chronic hepatitis B with standard-dose interferon-alfa monotherapy, use of higher-dose interferon-alfa for retreatment improves response (665,691,692). Lamivudine has also been used as secondary therapy for children without HIV infection who have not responded to standard interferon-alfa therapy (CII) (693--695); in HIV-infected children, initiation of a lamivudine-based HAART regimen could be considered (CIII).
For HIV/HBV-coinfected children developing lamivudine resistance during therapy, treatment options are more limited because of the lack of data on use of adefovir, entecavir, and tenofovir in children. Because these HBV drug-resistant isolates may have lower replicative capacity than wild-type HBV, some experts recommend continuing lamivudine or emtricitabine therapy in such cases. Alternatively, the addition of interferon-alfa therapy could be considered or, in children old enough to receive adult doses of tenofovir or adefovir, addition of tenofovir or adefovir to the regimen could be considered (CIII).
Prevention of Recurrence
Not applicable.
Discontinuing Secondary Prophylaxis
Not applicable.
Hepatitis C Virus
Epidemiology
In the United States, the prevalence of hepatitis C virus (HCV) infection is 0.2% among children aged 1--11 years and 0.4% among adolescents aged 12--19 years (696,697). The prevalence of HCV infection among HIV-infected children may be higher. In a serostudy of 535 HIV-infected children followed in pediatric HIV clinical trials, the prevalence of HCV infection by HCV antibody and RNA testing was 1.5% (698). In a more recent study of 228 HIV-infected children at an inner-city hospital in the Bronx, seven HIV-infected children had chronic HCV infection (3.1% [95% CI: 1.4%--6.5%]), defined as a reactive HCV antibody and positive HCV real-time PCR (646). The mean age of HIV/HCV-coinfected children was 16 years, and 57% had mild elevation (up to twofold above upper limit of normal) in serum transaminase levels.
Mother-to-child transmission is the predominant mode of HCV acquisition in children (699,700). Other potential sources of HCV infection in older children include injection-drug use, body piercing, tattoos, unintentional needlestick injury, household contact, and sexual exposure (701,702). Before 1992, blood transfusion was a source of HCV infection in children; a recent retrospective study found that 3% of infants who had received blood transfusions in a neonatal intensive-care unit during 1975--1992 were anti-HCV-positive (703). However, the incidence of HCV infection from transfusion has dramatically declined since 1992, when second-generation HCV EIA screening was implemented. With the current additional use of nucleic acid amplification testing, the risk for HCV infection through transfusion is approximately 1 per 2 million (704).
The overall risk for mother-to-infant transmission of HCV from a woman infected with HCV alone ranges from 4% to 10% (699,705--712). The primary risk factor for perinatal HCV transmission is maternal HCV viremia at delivery, although an absolute threshold for HCV transmission has not been identified (706,713--717). Data do not indicate that HCV genotype is related to risk for perinatal HCV transmission (706,712). Although a few studies have suggested that vaginal delivery increases risk for HCV transmission (705,707,709,713) and that HCV can be transmitted during the intrapartum period (718), most studies have found mode of delivery does not appear to influence perinatal HCV transmission (700,707,708,710,719--722). Additionally, even though HCV can be detected in breast milk, studies of infants born to HCV-infected women have not demonstrated a higher HCV transmission risk for breast-fed infants than for formula-fed infants (700,705--708,710,716,718,719,723).
Maternal HIV coinfection increases the risk for perinatal transmission, with perinatal HCV transmission rates of 6%--23% reported for infants born to women coinfected with HCV and HIV (699,705--707,711,717,720--722,724--729). Furthermore, a few studies suggest that children who acquire perinatal HIV infection may be more likely than children who did not acquire HIV to acquire HCV infection from HIV/HCV-coinfected mothers (720,721,727,729). Reported dual virus transmission has been reported in 4%--10% of children born to HIV/HCV-coinfected mothers (705,720,725,727,728). HCV RNA levels are hypothesized to be higher among women coinfected with HIV than women infected with HCV alone, which could account in part for the increased risk for mother-to-child HCV transmission from HIV/HCV-coinfected women; however, not all studies have found higher levels of HCV viremia among HIV-infected mothers (715,721,724). One European study suggested that HCV perinatal transmission may be reduced among HIV-infected women receiving HAART (722).
Chronic HCV infection, defined as the presence of HCV RNA for >6 months, appears to spontaneously resolve in 15%--40% of adults (730). Findings from a limited number of longitudinal studies suggest that HCV infection resolves spontaneously in 17%--59% of children with perinatal HCV infection (731--736). Viral clearance occurs by age 3 years in most children and may be more frequent in children infected with HCV genotype 3 (735,737).
Clinical Manifestations
Children with perinatal HCV infection appear to have a more benign clinical course than do adults with newly acquired HCV infection (701,738,739). Most HCV-infected children are asymptomatic, with minor abnormalities, such as hepatomegaly, or mild nonspecific symptoms, such as fatigue, myalgias, and poor weight gain (701,739,740); however, intermittent asymptomatic elevations in transaminase levels are common during the first 2 years of life (734,740,741). In a large European cohort of HCV-infected children, about 20% of children had apparent clearance of HCV viremia; 50% had chronic asymptomatic infection, characterized by intermittent viremia, rare hepatomegaly, and usually normal liver transaminase levels; and 30% had chronic active infection with persistent viremia and abnormal transaminase levels (735).
Histopathologic inflammatory changes of chronic hepatitis may be present in persons with chronic HCV infection despite lack of symptoms, normal serum transaminase levels, and low HCV RNA levels (740). However, most children with chronic HCV infection who have undergone liver biopsy and are included in published studies typically have mild-to-moderate liver disease as determined by signs of structural alterations, inflammatory activity, and necrosis (701,715,739,741). A small subset of children may develop more severe liver disease. In a study of 60 children with perinatally acquired or transfusion-acquired HCV infection who were infected for a mean duration of 13 years, 12% had significant fibrosis on liver biopsy (739). Older age at time of infection and elevated serum gamma-glutamyltranspeptidase correlated with fibrosis; serum transaminase levels correlated with inflammation (739).
In HIV/HCV-coinfected adults, the natural history of HCV infection appears to be accelerated, with more rapid progression to cirrhosis, decompensated liver disease, HCC, and death (742,743). Data are minimal on the effect of HIV/HCV coinfection on the natural history of HCV infection in children and are insufficient to draw conclusions about HCV disease progression in coinfected children (699).
Data on the impact of HCV infection on HIV disease progression in adults conflict; some studies suggest higher rates of HIV progression, and others do not (699). The effect of pediatric coinfection on HIV disease progression also is unclear because the number of coinfected children is small, and few studies have evaluated this. Two studies of children with perinatal HIV/HCV coinfection found no increase in HIV progression, but a study of older HIV/HCV-coinfected children with thalassemia infected through transfusion observed more rapid disease progression and higher mortality than in children infected only with HIV (720,728,744).
Diagnosis
Testing for HCV infection should be considered for any child whose mother is known to have HCV infection. All HIV-infected adults or adolescents should be tested for HCV infection.
Serologic and nucleic acid tests are used to diagnose HCV infection. HCV RNA first becomes detectable 1--3 weeks after HCV infection and precedes serologic response to HCV (730). A third-generation EIA is available for detecting antibody to HCV (anti-HCV). Passively transferred maternal anti-HCV can be detected for up to 18 months in infants born to HCV-infected mothers. In a large cohort of HCV-exposed but HCV-uninfected children, anti-HCV was present in 15% of children at 12 months, 5% at 15 months, and 2% at 18 months (715). Therefore, only the presence of persistent HCV viremia can be used to reliably verify HCV infection in at-risk infants aged <18 months (745). HCV infection can be diagnosed in such infants using a nucleic acid test to detect HCV RNA after 1 month of age; the sensitivity of the HCV RNA testing is low at birth (22%), but increases to 85% at 6 months (746). Most children with perinatal HCV infection will have a positive HCV RNA test by age 12 months. However, because of intermittent viremia, a single negative HCV RNA test is not conclusive evidence of lack of infection, and HCV RNA should be tested on at least two occasions between age 2 months and 6 months to definitively exclude HCV infection in an HCV-exposed infant (746).
A positive anti-HCV test in a child aged >18 months indicates HCV infection. Supplemental testing with a more specific assay, such as HCV RNA testing, is recommended to prevent the reporting of a false-positive result. A positive HCV RNA test confirms HCV infection and, if positive for >6 months, suggests chronic infection. HCV RNA can be measured qualitatively or quantitatively. Qualitative nucleic acid tests include qualitative PCR and transcription-mediated amplification. Quantitative tests include branched-chain DNA amplification, quantitative PCR, and real-time PCR and are most useful for monitoring response to anti-HCV therapy (730). Quantitative HCV RNA level (i.e., HCV viral load) does not correlate with degree of liver damage and does not serve as a surrogate for measuring disease severity, but it does provide important information about the response to antiviral therapy. Assays vary substantially, and if serial values are required to monitor antiviral therapy, continued use of the same quantitative assay for all assessments is strongly recommended.
Liver biopsy is the most accurate test to assess the severity of hepatic disease and quantitate the amount of hepatic fibrosis present. A liver biopsy may be useful for determining whether to initiate therapy for chronic HCV infection (652,657). However, liver biopsy is not required before the initiation of anti-HCV therapy, particularly in patients with a high probability of responding to therapy.
At least six HCV genotypes are known, with genotype 1 occurring most commonly in the United States (730). Persons with HCV genotypes 2 and 3 are more likely than those with genotype 1 to achieve sustained virologic response to anti-HCV therapy; thus, results from a liver biopsy may be less likely to affect decisions about the need for treatment in patients with genotypes 2 or 3 (657,687).
Prevention Recommendations
Prevention of Exposure
All HIV-infected persons should be screened for HCV. No reliable strategy exists to prevent perinatal HCV transmission. Cesarean delivery is not associated with reduced perinatal transmission of HCV infection and is not recommended for this purpose for women with chronic HCV infection who are HIV-uninfected. Scheduled cesarean delivery is recommended for HIV-infected women who have HIV RNA levels >1000 copies/mL near delivery to prevent perinatal HIV transmission (AII). Limited data suggest that breast-feeding does not transmit HCV. However, to prevent HIV transmission in the United States, where safe infant formula is available, HIV-infected women should not breast-feed (AII) (747).
No vaccines are available to prevent HCV infection. Adolescents considering tattooing or body-piercing should be informed about potential risks for acquiring HCV, which could be transmitted if equipment is not sterile or if proper infection-control procedures are not followed, and to avoid injection-drug use and unprotected sex (BIII) (748). HCV-infected persons should be advised not to share toothbrushes, razors, and other personal-care articles that might be contaminated with blood to prevent transmission of HCV.
Preventing First Episode of Disease
Patients with chronic liver disease can develop fulminant hepatitis from hepatitis A or B infection; all children (regardless of HIV and HCV infection status) should receive standard vaccination with hepatitis A and B vaccines (AIII) (39,40,748).
Treatment Recommendations
Treatment of Disease
Published studies are limited on treatment recommendations for HCV-infected children. Current pediatric trials in the United States---including the PEDS-C study, a randomized, double-blind, placebo-controlled trial of pegylated interferon-alfa with and without ribavirin---should provide additional data. Data on treating children coinfected with HCV and HIV are even more limited. Consultation with experts in treating chronic HCV infection in children is recommended.
HIV/HCV-coinfected adults and adolescents
Treatment should be considered in any nonpregnant HCV-infected adult, regardless of HIV coinfection status, who has abnormal serum transaminase levels with a liver biopsy showing chronic hepatitis with significant fibrosis and compenstated liver disease (749). Treatment should be considered for HIV/HCV-coinfected adults for whom potential benefits of treatment are judged to outweigh potential risks, including those infected with HCV genotype 2 or 3, those with stable HIV infection not requiring antiretroviral therapy, and those with cryoglobulinemic vasculitis or glomerulonephritis (652,750). Baseline serum HCV RNA level and HCV genotype are the primary predictors of response to treatment; younger age, higher CD4 count, elevated transaminase levels, lack of liver fibrosis, low body mass index, lack of insulin resistance, and white race are other variables associated with better treatment response (750). The recommended treatment is combined pegylated-interferon-alfa-2a or-2b plus daily oral ribavirin for 48 weeks regardless of HCV genotype. In HIV/HCV-coinfected adults, rates of sustained virologic response range from 44% to 73% for treatment of HCV genotype 2 and 3 infection and from 14% to 29% for HCV genotype 1 infection (641,751,752). Although some preliminary data from clinical treatment trials suggest better rates of sustained virologic response, these may result from better preselection of patients for therapy or improved adherence after dose adjustment(s). Response to anti-HCV treatment improves in HIV/HCV-coinfected adults with CD4 count >200 cells/mm3; therefore, HAART should be considered before anti-HCV therapy is initiated in HIV/HCV-coinfected patients with CD4 count <200 cells/mm3. Anti-HCV treatment usually is not recommended during pregnancy for HCV-infected women because ribavirin is teratogenic.
HCV-infected children without HIV infection
Treatment of HIV-uninfected children aged <3 years who have HCV infection usually is not recommended because spontaneous HCV clearance can occur in this age group (DIII). All decisions about treatment of HCV infection in children should be individualized because HCV usually causes mild disease in children, and few data exist to identify risk factors differentiating those at greater risk for progression of liver disease (641,753).
The only currently FDA-approved therapy for HCV-infected children aged 3--17 years with compensated liver disease is combined standard interferon-alfa-2b and ribavirin. Standard interferon-alfa is administered by subcutaneous injection three times per week. Ribavirin oral solution has been approved for treatment of chronic HCV infection among children aged ≥3 years. For HIV-uninfected children with HCV infection, a 24-week course of therapy is recommended for genotypes 2 and 3; 48-week courses are administered for other HCV genotypes. Combination therapy with standard interferon-alfa and ribavirin results in overall rates of sustained virologic response of 46%--65% and is well-tolerated in children (657,687,754--757). Similar to adults, children infected with genotype 1 were less likely to have a sustained virologic response (36%) than those infected with genotype 2 or 3 (84%) (687). Other factors associated with favorable response to anti-HCV treatment in children include lower pretreatment HCV RNA levels, white race, and possibly younger age (657).
HIV/HCV-coinfected children
No specific treatment studies have been done of children with HIV/HCV-coinfection, and recommendations are based primarily on data from adults. Because therapy for HCV infection is more likely to be effective in younger patients and in those without advanced disease or immunodeficiency, treatment should be considered for all HIV/HCV-coinfected individuals, including HIV-infected children aged >3 years who have no contraindications to treatment (BIII). Some specialists would treat children infected with HCV genotypes 2 or 3 without first obtaining a liver biopsy (BIII). Pegylated interferon-alfa, which is administered by injection once weekly for 48 weeks combined with ribavirin, is recommended for treatment of HCV infection in adults. However, pegylated interferon-alfa is not FDA-approved for use in HCV-infected children---although it is under study. On the basis of the increased efficacy of combination therapy with ribavirin and either standard or pegylated interferon-alfa and on data from adults, treatment of HCV-infected children, regardless of HIV status, should include combination therapy with ribavirin and interferon-alfa (BIII). In HIV/HCV-coinfected adults, the recommended duration of treatment is 48 weeks for infections with all HCV genotypes, including 2 and 3, because coinfected adults may not respond as well as those without HIV infection and may have greater relapse rates. Moreover, the efficacy of shorter treatment duration has not been adequately evaluated in HIV-infected persons (750). By extrapolation, 48 weeks of therapy also are recommended for HIV/HCV-coinfected children (BIII). Potential drug interactions complicate the concomitant use of antiretroviral therapy and anti-HCV therapy. Ribavirin enhances phosphorylation of didanosine, which could increase the risk for toxicity; therefore, these drugs should not be used together (EIII). Ribavirin and zidovudine both are associated with anemia and, when possible, should not be administered together (DII) (750).
Monitoring and Adverse Events, Including IRIS
Although no evidence-based long-term monitoring guidelines exist for children with perinatally acquired HCV, many experts monitor HCV RNA levels and serum transaminase levels every 6--12 months and hemogram and serum alpha fetoprotein levels annually (748). Serum transaminase levels can fluctuate and do not necessarily correlate with histologic liver damage because significant liver disease can be present in patients with normal serum transaminase levels. In HCV-infected persons without HIV, HCC rarely is seen in the absence of cirrhosis. The benefits of serum alpha fetoprotein and abdominal sonography as screening tools for HCC have not been studied in children. Some experts will perform periodic sonographic screening at defined intervals (every 2--5 years) in children with chronic HCV infection; others will do these tests only in those with advanced liver disease and/or rising serum alpha fetoprotein concentrations (748). The risk for HCC in HCV-infected children, with or without HIV infection, is not known.
HCV RNA quantitation is used to monitor response to antiviral therapy. HCV RNA levels should be performed at baseline, after 12 and 24 weeks of antiviral therapy, at treatment completion (48 weeks), and 6 months after treatment cessation. Some experts continue to perform serial HCV RNA testing at 6- to 12-month intervals for an additional 1--5 years to exclude late virologic relapse. Decreases in HCV RNA ≥2 logs below the baseline during the first 12 weeks of therapy constitute an early virologic response (730). A sustained virologic response is defined as absence of detectable HCV RNA using an HCV RNA assay with a lower limit of detection of ≥50 IU/mL at 24 weeks after antiviral treatment ends. Relapse is defined as HCV RNA rebound at the end of therapy after an initial response to undetectable HCV RNA levels. Nonresponse is defined as failure to suppress HCV RNA below detection at any time during treatment; breakthrough is the reemergence of detectable HCV RNA after suppression below the limits of detection despite the continuation of therapy.
In the absence of data from HIV/HCV-coinfected children, the same criteria for determining response to therapy in HIV/HCV-coinfected adults should be used. If an early virologic response is observed after the first 12 weeks of treatment, completion of additional HCV therapy is recommended. Adults who do not achieve an early virologic response by week 12 have a limited chance (<3%) of achieving sustained virologic response, regardless of duration of therapy, and treatment can be discontinued after 12 weeks in such patients. Persons who achieve a ≥2 log10 reduction in HCV RNA level but who have detectable HCV RNA after 12 weeks of therapy should be retested after completion of 24 weeks of therapy. If HCV RNA remains detectable after 24 weeks, anti-HCV treatment should be stopped, whereas an additional 24 weeks of therapy is indicated (total 48 weeks) if HCV RNA is not detected at that time. Persons who achieve an undetectable HCV RNA level after 12 weeks of therapy should complete an additional 36 weeks of anti-HCV treatment (total 48 weeks).
In addition to HCV RNA quantitation, patients receiving antiviral therapy for HCV infection should be closely monitored for medication side effects with complete blood count, serum transaminase levels, and tests of thyroid function. If the child also is initiating HAART, some experts would monitor transaminase levels more frequently during the first few months of therapy (e.g., monthly for 3 months) because of the risk for IRIS (see below).
Side effects of interferon-alfa in children, although frequent, usually are not severe; approximately 5% of children require treatment discontinuation because of side effects. The most common side effects include influenza-like symptoms (e.g., fever, chills, headache, myalgias, arthralgias, abdominal pain, nausea, and vomiting) in 80% of patients during the first month of treatment. However, these symptoms usually resolve over time and usually are not treatment-limiting; premedication with acetaminophen or ibuprofen might reduce the incidence of side effects. Subtle personality changes that resolve when therapy is discontinued have been reported in 42% of children (686). Depression and suicidal ideation also have been reported in clinical trials of children treated with interferon-alfa (687). Neutropenia, which resolves after discontinuation of therapy, is the most common laboratory abnormality; anemia and thrombocytopenia are less common. Abnormalities in thyroid function (hypothyroidism or hyperthyroidism) have been reported with interferon-alfa therapy (688). Loss of appetite, with transient weight loss and impaired height growth, can occur but usually resolves after completion of therapy (689). Less commonly observed side effects of interferon-alfa include epistaxis and transient mild alopecia. Certain children have developed antinuclear autoantibodies. Interferon-alfa therapy is contraindicated for children with decompensated liver disease; substantial cytopenias; severe renal, cardiac, or neuropsychiatric disorders; and autoimmune disease (EII) (690).
Side effects of ribavirin include hemolytic anemia and lymphopenia. Ribavirin-induced hemolytic anemia is dose-dependent and usually presents with a substantial decrease in hemoglobin within 1--2 weeks after ribavirin initiation, but the trend usually stabilizes. Significant anemia (hemoglobin <10 g/dL) occurs in about 10% of ribavirin-treated children (753). Use of erythropoietin for managing clinically significant anemia during HCV treatment can be considered (BIII). Coadministration of didanosine is contraindicated in children receiving ribavirin because this combination can increase the risk for mitochondrial toxicity and hepatic decompensation (EIII). Children receiving concomitant zidovudine may be more likely to experience bone marrow suppression; if possible, zidovudine should be avoided in children receiving ribavirin (DIII). If zidovudine and ribavirin are administered together, the child should be monitored closely for neutropenia and anemia. Ribavirin is teratogenic and should not be used in pregnant women. Sexually active adolescent boys and girls or those likely to become sexually active who are receiving ribavirin should be counseled about the risks and need for consistent contraceptive use during, and for 6 months after completion of, ribavirin therapy.
As with HIV/HBV coinfection, the institution of HAART in HIV/HCV-coinfected patients can worsen hepatitis, with increases in serum transaminase levels and clinical signs of liver disease, including hepatomegaly and jaundice. This does not represent a failure of HAART but rather a sign of immune reconstitution. IRIS manifests by an increase in serum transaminase levels as the CD4 count increases during the first 6--12 weeks of HAART. Thus, serum transaminase levels should be monitored closely after introduction of HAART in HIV/HCV-coinfected children. The prognosis for most persons with IRIS is favorable because a robust inflammatory response may predict an excellent response to HAART in terms of immune reconstitution and, perhaps, improved survival. In a patient experiencing a hepatic flare, differentiating between IRIS and drug-induced liver toxicity may be difficult, and no reliable clinical or laboratory predictors exist to distinguish between the two. Close interaction of the HIV specialist with a specialist in hepatic disease is recommended for such patients; prompt consultation with a hepatologist should be sought if elevated aminotransferases are associated with clinical jaundice or other evidence of liver dysfunction (e.g., low serum albumin).
Management of Treatment Failure
No data exist on which to base recommendations for treatment of HIV/HCV-coinfected children or adults in whom initial HCV treatment fails. In HIV/HCV-coinfected adults, a second course of treatment for nonresponders (those who do not achieve early virologic response by week 12 or undetectable HCV load at week 24) or patients whose HCV relapses has limited chances of resulting in sustained virologic response. Therapeutic interventions for such patients need to be individualized according to prior response, tolerance, and adherence to therapy; severity of liver disease; viral genotype; and other underlying factors that might influence response. Some experts might extend the duration of treatment (e.g., to 72 weeks) for adults who experience a virologic response followed by relapse after adequate HCV therapy or for patients with advanced fibrosis, long-term administration of low-dose pegylated interferon. No data exist for HIV/HCV-coinfected children on which to base a recommendation.
Prevention of Recurrence
Not applicable.
Discontinuing Secondary Prophylaxis
Not applicable.
Human Herpesvirus 6 and 7
Epidemiology
Human herpesvirus 6 (HHV-6) and herpesvirus 7 (HHV-7) are closely related members of the Roseolovirus genus of herpesviruses. Humans are the only known natural host for these two viruses. The cellular reservoir is believed to be peripheral blood mononuclear cells (PBMCs) and possibly epithelial cells of the salivary glands and the bronchial tree. The infection is believed to be transmitted primarily through saliva; sexual transmission may occur, and presumptive in utero infection has been described. Reactivation, with salivary shedding, occurs frequently among immunocompetent children and adults (758).
HHV-6 has two subtypes, A and B. HHV-6B is the etiologic agent of most primary infections; HHV-6A has been linked to neurologic syndromes. The vast majority of children become infected with HHV-6 early in childhood, with 70%--90% of children HHV-6 seropositive by age 2 years, and virtually 100% by age 3 years (759,760). In a large prospective study of North American children, the peak age of acquisition of HHV-6 was 6--9 months (761). Such infection is associated with varied clinical manifestations, viremia, and the frequent persistence of the viral genome in PBMCs (762).
HHV-6 also can be transmitted from mother to child. In several recent studies, virtually 100% of pregnant women were HHV-6--seropositive. During pregnancy, 2%--9% of women shed virus in the genital tract (763,764). Congenital HHV-6 infection has been documented in ≤2% of newborns (760,761,765,766). In one large study of approximately 5000 births, 1% of cord blood samples were positive for HHV-6 DNA. None of the congenitally infected infants were symptomatic (761). The virus does not appear to be transmitted by breast-feeding.
HHV-6 infects the same target cells as HIV, and HHV-6 and HIV can simultaneously infect the same CD4 cells under experimental conditions. Some evidence exists for interactions between HHV-6 and HIV at the cellular or molecular levels. Some studies have suggested that HHV-6 upregulates HIV expression in coinfected cells in vitro (767,768).
In a study of HHV-6 infection in 227 children born to HIV-infected mothers, three (7%) of 41 HIV-uninfected infants were positive for HHV-6 DNA in the first month of life, suggesting possible intrauterine infection (759). The cumulative infection rates of HHV-6 at ages 6 and 12 months were significantly lower in HIV-infected children (11% and 33%, respectively) than in HIV-uninfected children (28% and 78%, respectively), and high CD4 percentage (>15%) in the child before HHV-6 infection was associated with high HHV-6 infection rate. However, HIV disease progressed longitudinally in all 10 infants with HIV/HHV-6 coinfection and in 58% of those without HHV-6 coinfection, suggesting that HHV-6 coinfection might increase risk for HIV disease progression.
HHV-7 is acquired later in life than HHV-6. In contrast to HHV-6, seropositivity to HHV-7 is approximately 50% at age 2 years. Reactivation and/or persistent shedding also can occur. Salivary shedding is common, noted in up to 90% of seropositive adults, and HHV-7 DNA has been found in breast milk (unlike HHV-6) (769).
Clinical Manifestations
Many cases of primary infection with HHV-6, which usually occurs by age 2 years, are asymptomatic or accompanied by mild, nonspecific symptoms. The most common symptom is fever, which can be high and abrupt, associated with crankiness and rhinitis. In several studies, primary HHV-6 infection was associated with infants presenting for care of acute febrile illness. Among children aged <3 years presenting for evaluation of a febrile illness, 10% had primary HHV-6 infection; the incidence was 20% for febrile infants aged 6--8 months (770).
HHV-6 is the causative agent of most cases of exanthem subitum (also known as roseola infantum), a febrile illness of early childhood, associated with a distinctive exanthem. The incidence of HHV-6--related exanthem subitum peaks at age 6--9 months and is associated with fever of 4--5 days, with rash developing as the fever subsides (771). Ten percent to 20% of infants with primary HHV-6 infection will present with exanthem subitum. HHV-7 also has been associated with exanthem subitum.
Both primary infection with and reactivation of HHV-6 has been associated with several CNS syndromes in immunocompetent children and adults, including febrile and nonfebrile seizures, encephalitis/encephalopathy and acute necrotizing encephalopathy (770,772,773). Convulsions, a feature of severe HHV-6 infection, often can be prolonged and complicated (774).
Reactivation of HHV-6 occurs in adults with immunodeficiency or in patients on immunosuppressive therapy after transplantation. Although the exact frequency of HHV-6 reactivation in transplant recipients (bone marrow and solid organ) is difficult to determine, it appears to be common and occurs within the first 12 weeks after transplantation (775). Most reactivation involves HHV-6B. Many episodes of reactivation are asymptomatic, but when symptoms occur, they can include fever, skin rash, and leukopenia. Pneumonitis, encephalitis, bone marrow suppression, and graft versus host disease have been associated with reactivation of HHV-6 after bone marrow transplantation (775,776).
HIV-infected persons exhibit frequent reactivation of HHV-6 (775); whether this causes symptoms or progression of illness is controversial. Symptoms most likely to be associated with HHV-6 are pneumonitis and encephalitis.
Reactivation of HHV-7 also occurs in immunocompetent and immunodeficient persons. The relation of HHV-7 reactivation to disease states is still poorly understood.
Diagnosis
Many primary infections result in a nonspecific febrile illness. The 10%--20% of primary infections that manifest as exanthem subitum can be clinically diagnosed on the basis of symptomatology and distinctive rash.
Diagnosing HHV-6--related illness can be difficult because of the frequent reactivation in immunocompetent healthy children and adults. Specific testing for HHV-6 or -7 infection may be performed using such laboratory methods as serology, culture, antigen detection, PCR, in situ hybridization, and immunohistochemistry. The latent nature of these herpesviruses must be considered, and the differing capability of these tests to distinguish nonreplicating latent virus from replicating active virus must be understood (777). Laboratory evaluations (e.g., serology and PCR) to diagnose HHV-7 infections are rarely used and typically limited to research.
Demonstration of an HHV-6--specific antibody seroconversion or significant change in antibody titer between acute and convalescent paired serum samples can diagnose infection with HHV-6; delay can occur between initial infection and seropositivity, especially in immunodeficient patients. The sensitivity of serology varies and does not distinguish between HHV-6A and HHV-6B. Some cross reactivity may occur with HHV-7.
Identification of HHV-6 or -7 in PBMCs by virus culture firmly establishes the presence of active infection in immunocompetent hosts, but association with specific disease is problematic in immunocompromised patients because of a low background rate of viremia. Virus culture requires cocultivation with PBMCs or cord blood and requires 1--3 weeks (777). Therefore, it is available only in specialized research laboratories.
PCR can be used to detect HHV-6 DNA or RNA. A positive serum, plasma, or PBMC viral DNA or RNA PCR assay, in the absence of measurable antibody, indicates a primary infection. HHV-6 is found in cellular material such as PBMCs and tissue even during latency; therefore, after primary infection a positive DNA PCR cannot be used to establish reactivation, whereas detection of cell-free viral DNA or RNA in plasma, serum, or CSF by PCR (776) is considered evidence of active viral replication---and thus reactivation---in a seropositive patient. Evidence of reactivation and clinical disease must be correlated cautiously because of the frequent asymptomatic reactivation in most adults and children (775,776).
Prevention Recommendations
Preventing Exposure
Because HHV-6 and HHV-7 infections are ubiquitous, prevention of primary infection is not possible. Among transplant recipients, prophylactic ganciclovir may decrease the number of episodes and severity of HHV-6 reactivation (778,779).
Preventing First Episode of Disease
Given the ubiquity of HHV-6 and -7 during early childhood and the lack of an effective vaccine, prevention of HHV-6 disease is not feasible.
Discontinuing Primary Prophylaxis
Not applicable.
Treatment Recommendations
Treatment of Disease
Most HHV-6 primary infections result in a mild, self-limited febrile illness. For the immunodeficient adult or child with possible HHV-6--associated lung or CNS disease, care must be used to exclude other diagnostic possibilities. No clear indications exist for treating HHV-6 infection in HIV-infected children, although treatment might be considered for the rare instance of severe encephalitis proven to result from HHV-6 (CIII). However, no clinical trials or proven therapies exist for HHV-6. On the basis of data in adults, drugs that might be considered for severe HHV-6 disease are ganciclovir, foscarnet, and cidofovir. However, even though in vitro data suggest ganciclovir and foscarnet are active against HHV-6, data are limited that support their use among HIV-infected patients with possible HHV-6 related illness (CIII) (776,780,781). Ganciclovir has been used to treat HHV-6 encephalitis in adult transplant patients (782). However, limited success of ganciclovir therapy in preventing fatal outcome has been reported; in the patients who died, ganciclovir did not reduce HHV-6 load in CSF (783). Case reports have documented both successful and disappointing results of foscarnet treatment for HHV-6 encephalitis in transplant recipients (783--785). Cidofovir followed by foscarnet has been used in a stem cell transplant recipient who developed HHV-6 encephalitis with evidence of a significant reduction in HHV-6 load in CSF and in plasma after cidofovir administration (786). One case of successful use of high-dose ganciclovir to treat HHV-6 encephalitis was reported in a pediatric bone marrow transplant patient (787). Given the lack of data in children, no specific recommendations can be made.
HHV-7 has not been recognized as responsible for any specific disease in HIV-infected persons, and no treatment is indicated.
Monitoring and Adverse Events, Including IRIS
The major side effect of ganciclovir is myelosuppression (i.e., anemia, neutropenia, and thrombocytopenia). Dose reduction or interruption because of hematologic toxicity may be necessary in ≤40% of patients receiving IV ganciclovir; granulocyte colony-stimulating factor can be used to ameliorate marrow suppression. The main toxicities of foscarnet are decreased renal function and metabolic derangements. For patients receiving ganciclovir or foscarnet, complete blood counts, serum electrolytes, and renal function should be monitored twice weekly during induction therapy and once weekly thereafter. The major side effect of cidofovir is potentially irreversible nephrotoxicity; the drug produces proximal tubular dysfunction, including Fanconi syndrome and acute renal failure. When present, renal toxicity manifests as proteinuria and glycosuria. To minimize nephrotoxicity, probenicid should be administered before each infusion, and IV hydration with normal saline should be administered before and after each cidofovir infusion. For patients receiving IV cidofovir, blood urea nitrogen, creatinine, and urinalysis should be performed before each infusion; administration of the drug is contraindicated if renal dysfunction or proteinuria is detected. Other reported adverse events include anterior uveitis and ocular hypotony; serial ophthalmologic monitoring for anterior segment inflammation and intraocular pressure is needed while the drug is being received systemically. Cidofovir should not be administered concomitantly with other nephrotoxic agents. Cidofovir therapy must be discontinued if serum creatinine increases ≥0.5 mg/dL above baseline.
HHV-6 and HHV-7 have not been associated with IRIS with HAART treatment in HIV-infected children or adults.
Management of Treatment Failure
Mutations conferring resistance of HHV-6 to ganciclovir, cidofovir, and foscarnet have been described (788). Whether a change from one drug to the other would be beneficial is unknown.
There are currently no recommendations for the management of treatment failure.
Prevention of Recurrence
No data exist on preventing HHV-6 or HHV-7 reactivation from latency in HIV-infected patients.
Discontinuing Secondary Prophylaxis
Not applicable.
Human Herpesvirus 8 Disease
Epidemiology
Human herpesvirus 8 (HHV-8) is a transmissible DNA virus, with similarities in DNA structure to Epstein-Barr virus. HHV-8 has been causally linked to all forms of KS (HIV-related and endemic KS) and with two rare neoplastic conditions usually associated with HIV infection, body cavity-based lymphoma and multicentric Castleman disease. The exact mechanism by which HHV-8 infection leads to neoplastic disease has not been fully elucidated, but seroconversion to HHV-8 antibody positivity virtually always precedes development of the tumors (789). Higher plasma HHV-8 DNA titers are associated with increased risk for KS (790).
The prevalence of antibodies to HHV-8 varies widely with age and geography. In the United States and Europe, 1%--3% of the general adult population is seropositive, with higher rates (8%) among men who have sex with men (791). Among other adult men in the general population, HHV-8--seropositivity was marginally associated with duration of heterosexual activity and positively associated with the number of lifetime sex partners and coinfection with HBV and HSV type 2 viruses; none of these were significantly associated with risk for women. In contrast, the seropositivity rate in some areas of Africa is >80% (792--795).
HHV-8 is transmitted through oral and genital secretions. Immunocompetent HHV-8--infected adults frequently shed HHV-8 in their oropharyngeal secretions, with virus detected in saliva on 22% of test days (796). In areas where HHV-8 infection is endemic, the seroprevalence increases quickly during the first 5 years of life, especially when other family members are HHV-8--positive, then plateaus until adolescence and young adult years. In a study in rural Tanzania, the rate of positivity for HHV-8 was 3.7% among infants, 58% among children aged 4--5 years, and 89% among adults aged >45 years (797). The incidence among infants and children increased with the number of HHV-8-positive parents and siblings in the home, indicating nonsexual transmission for prepubertal children, with a limited role for perinatal transmission (797--803).
In the United States, among a cohort of high-risk HIV-infected and HIV-negative adolescents, with a median age of 19 years, 11.2% were HHV-8--positive (804). The highest rates were in adolescent HIV-infected males reporting sex with males (23%). Seropositivity was associated with HIV infection, men who have sex with men, a history of syphilis, and injection-drug use (804,805).
HHV-8 also may be transmitted through exposure to infected blood. Adult injection-drug users have an increased rate of HHV-8 positivity (804,805). In addition, recent evidence suggests that HHV-8 may be transmitted through blood product transfusions. In one study in an area of Uganda with a high incidence of HHV-8--seropositivity, the excess risk for acquiring HHV-8 through transfusion was nearly 3%, recipients of HHV-8 antibody-positive blood were compared with those receiving HHV-8--negative blood (806).
A small study suggested that maternal HHV-8 infection might increase risk for perinatal transmission of HIV, although no evidence of HHV-8 infection was identified among HIV-infected infants (801). Women coinfected with HHV-8 and HIV had increases in both HHV-8 and HIV viral load in serum and/or cervical fluid during pregnancy (807).
During the pre-HAART era, the overall incidence of KS among HIV-infected adults was as high as 20%. However, the rate among children was low. In the United States and England, KS represented <1% of pediatric AIDS-defining illnesses.
The incidence of KS appeared to decline even before the widespread use of HAART. The reason for this decline was unclear but may have been related to the use of other antiviral agents, such as those used to treat CMV (i.e., foscarnet, ganciclovir, and cidofovir), which may inhibit HHV-8 (808--814). KS, primary effusion lymphoma, and multicentric Castleman disease are described most frequently among HIV-infected persons with more advanced immunosuppression (CD4 count of <200 cells/mm3), but they can occur at any CD4 count. With the advent of earlier and more aggressive HAART, the incidence of KS in adults has continued to decrease (815).
Although KS occurs primarily in adults, the incidence in children has increased substantially as a result of the HIV pandemic, particularly in Africa, and the frequent use of immunosuppressive drugs. One series reported a 40-fold increase in incidence of childhood KS in Uganda in the era of AIDS (816).
Clinical Manifestations
A febrile illness with mild respiratory symptoms and a maculopapular rash has been associated with primary infection in young immunocompetent children (817). A similar self-limited illness has been described in adults with primary infection. Evidence suggests more significant symptomatology among immunodeficient adults with primary infection, including reports of fever, arthralgia, splenomegaly, and bone marrow suppression (818,819).
KS presentation varies widely, but most persons have nontender, purplish, indurated skin lesions; intraoral lesions can be seen and visceral dissemination can occur, occasionally without skin lesions. Multicentric Castleman disease presents with generalized adenopathy and fever and may progress to multiorgan failure.
Diagnosis
The laboratory diagnosis of HHV-8 is most commonly based on serologic assays, such as immunofluorescence, enzyme-linked immunosorbent assay, and Western blot. However, without a standard for diagnosing HHV-8 infection, these tests range in sensitivity from 80% to ≥90% and demonstrate poor interassay agreement (820). Combination assays containing both lytic and late-phase antigens may improve detection rates. Nucleic acid-based tests, such as in situ DNA hybridization and PCR, are important for pathologic diagnoses in biologic specimens. Although these tests have high levels of sensitivity, specificity and reproducibility are highly variable. Routine screening for HHV-8 by PCR or serologic testing is not indicated for HIV-infected persons.
Prevention Recommendations
Preventing Exposure
For HIV-infected persons, coinfection with HHV-8 places them at risk for KS. The risk is highest in adults (compared with children) and in persons with severe immunodeficiency. Routine testing of children and adults for HHV-8 is not recommended; therefore, the serostatus of newly identified HIV-infected persons usually is not known. For adolescents in whom KS is diagnosed, counseling should include the possibility of transmitting HHV-8 to their sexual contacts through intercourse and possibly kissing. Although efficacy of condom use for preventing HHV-8 exposure has not been established, HIV-infected persons should use latex condoms during every act of sexual intercourse to reduce exposure to sexually transmitted pathogens. HIV-infected injection-drug users should be counseled not to share drug-injection equipment, even if both users are already HIV-infected, because of the risk of becoming infected with HHV-8 or other bloodborne pathogens.
In the future, HHV-8 testing of donated blood products, before use for immunodeficient patients, might be considered. In addition, the routine use of leukocyte reduction for red cell transfusions may lower the transmission risk.
Infants can acquire HHV-8 perinatally or through contact with infected family members and playmates. No effective intervention is known to prevent childhood acquisition of HHV-8.
Preventing First Episode of Disease
The use of HAART with suppression of HIV replication has markedly decreased the incidence of KS among HIV-infected adults and should be the goal of treatment wherever possible (AII). Routine testing to identify HHV-8 seropositive HIV-infected persons is not recommended (DIII). Although several antiviral agents inhibit HHV-8 replication in vitro (e.g., ganciclovir, foscarnet, cidofovir), no data exist on their use to prevent KS in persons coinfected with HIV and HHV-8.
Treatment Recommendations
Treatment of Disease
Because the HIV-related clinical entities associated with HHV-8, such as KS and Castleman's disease, are oncologic and traditionally have been treated with cytotoxic chemotherapy, specific treatment is not included in this report. However, effective suppression of HIV replication with HAART among HIV-infected patients with KS might prevent KS progression or occurrence of new lesions and should be considered for all persons with evidence of active KS and other HHV-8--associated malignant lymphoproliferative disorders (AII).
In HIV-infected adults, HHV-8 cellular viremia and higher viral load have been associated with disease progression (821). Treatment with specific antiviral agents, such as ganciclovir, foscarnet, and cidofovir, which have in vitro activity against the lytic but not latent phase of HHV-8, has not been widely studied. Additionally, the vast majority of infected cells are not undergoing lytic replication, and antiherpes medications have had little or no effect on established KS or HHV-8 cellular viremia. Efforts to induce lytic replication or to attack the episomal (latent) HHV-8 genome are in progress (822,823).
In contrast to KS, many of the cells in Castleman disease support lytic replication of HHV-8, and treatment of Castleman disease with antiherpesvirus drugs has led to substantial clinical improvement in some studies (823). The use of IV ganciclovir or oral valganciclovir is recommended for treating multicentric Castleman disease (BII) (824) and may be a useful adjunct for treating primary effusion lymphoma (BII) (825,826). Appropriate chemotherapy, in combination with potent antiretroviral therapy, should be considered for patients with visceral KS or primary effusion lymphoma (BII).
Monitoring and Adverse Events, IRIS
Rapid progression of KS after initiation of HAART and after a change from a failing regimen to a more potent one have been reported. Progression of KS, representing IRIS, usually appeared within 8 weeks after start of a potent HAART regimen. Most patients experienced rapid progression of cutaneous lesions; however, sudden worsening of pulmonary KS, with resultant deaths, was reported in at least four patients. All reported fatalities were linked to pulmonary KS. In most cases, HAART was continued with stabilization and then regression of lesions. In more severe cases, especially those involving visceral lesions, chemotherapy was instituted and, in combination with HAART, led to regression of the KS (827,828).
Management of Treatment Failure
No recommendations exist for management of treatment failure.
Prevention of Recurrence
Effective suppression of HIV replication with HAART among HIV-infected patients with KS might prevent KS progression or occurrence of new lesions and should be considered for all persons with evidence of active KS (BII).
Discontinuing Secondary Prophylaxis
Not applicable.
Herpes Simplex Virus
Epidemiology
HSV-1 and HSV-2 affect all populations. HSV-1 is transmitted primarily through contact with infected oral secretions; HSV-2 is acquired primarily through contact with infected genital secretions. In developed countries, urban poor populations acquire HSV-1 at a younger age (70%--80% seropositive by age 20 years) than do the rest of the population (30%--40%) (829). In a large U.S. study, HSV-1 seroprevalence in children aged 6--13 years was 31% and increased with age, from 26% in 6- to 7-year-olds to 36% in 12- to 13-year-olds, and varied by race/ethnicity, birthplace, and poverty levels (higher in non-Hispanic blacks, children born in Mexico, and children living below poverty level) (830). The seroprevalence of HSV-1 approaches 60% among the general older adult population (831). HSV-2 seroprevalence in persons aged 14--49 years was 17% and increased with age, from 2% in 14- to 19-year-olds to 26% in 40- to 49-year-olds. HSV-2 seroprevalence was higher in non-Hispanic blacks, persons with large numbers of sex partners, and females and varied by birthplace and poverty level (831,832).
An HSV-infected mother can transmit HSV to her infant, resulting in neonatal infection. In older children and adults, classic HSV transmission is person to person through direct contact with infected oral secretions or lesions. Neonatal HSV infection rate is 1 case per 2000--15,000 deliveries (833). Neonatal transmission occurs primarily through exposure of the infant to HSV-infected maternal genital fluids during passage through the birth canal; by ascending infection; or through use of invasive procedures, such as fetal scalp monitoring, that disrupt fetal skin integrity during labor (834). Congenital (in utero) HSV acquisition is rare but can result in devastating cutaneous, ocular, and CNS damage.
Maternal HSV antibody status before delivery influences both the severity and likelihood of transmission to the infant (834). The risk for neonatal HSV infection is greatest in an infant born to a woman with primary HSV infection (range: 30%--50%) (829). The risk is much lower (0%--5%) for infants born to women shedding HSV caused by reactivated infection (589). Genital shedding of HSV at delivery increases risk for transmission, and prolonged rupture of membranes (>6 hours) increases the risk, probably because of ascending HSV infection from the cervix. Cesarean delivery substantially lowers the risk for transmission (829,834,835). Importantly, many mothers of neonates with HSV-related illness will have pregnancy histories void of either past HSV infection or primary lesions (834). In the United States, 75% of neonatal infections are caused by genital herpes, HSV type 2, and the remainder by HSV type 1.
In a study of seroprevalence among pregnant U.S. women, overall HSV-1 seroprevalence was 63%; HSV-2 seroprevalence, 22%; and infection with both HSV-1 and 2, 13% (836). HSV seroprevalence differed by race/ethnicity and number of lifetime sex partners. HSV-2 infection rates might be higher in HIV-infected than HIV-uninfected women. Women infected with HIV, particularly those with low CD4 count, shed HSV from the vulva and cervix more commonly than do HIV-uninfected women; most of this shedding is asymptomatic (589,837). Among HIV-uninfected women, the rate of HSV reactivation is approximately 25% during the last month of pregnancy, but 2%--3% will be shedding on the day of delivery (838). In comparison, in women coinfected with HIV and HSV, an estimated 10% have cervical shedding of HSV on the day of delivery (839). The risk for genital HSV reactivation and shedding increases as HIV-related immunosuppression progresses (837,839). No evidence indicates that in utero HSV infection of the infant occurs more frequently in HIV-infected pregnant woman or whether infants born to women coinfected with HIV and HSV-2 are at increased risk for perinatal (intrapartum) HSV infection. However, HSV infection may increase the risk for mother-to-child HIV transmission. In a study in Kenya of women receiving zidovudine prophylaxis, the presence of HSV-2--related genital ulcers in late pregnancy was associated with increased plasma HIV RNA levels and increased risk for intrapartum HIV transmission, even after adjustment for plasma HIV RNA levels (840).
Recurrent or persistent HSV infection is the AIDS-indicator condition in approximately 6% of children with AIDS. In a study in HIV-infected children in the United States during the HAART era, the incidence of systemic HSV infection was 0.9 per 100 child-years and was most common among children with reduced CD4 percentage (<25%) (3). As in HIV-infected adults, HIV-infected children have more frequent and severe episodes of HSV reactivation. Five percent to 10% of children with AIDS and primary gingivostomatitis develop frequent recurrences, which can be associated with severe ulcerative disease and symptoms similar to primary infection (841). HIV-infected children also can shed virus longer with both primary and reactivation HSV infection than can HIV-uninfected children.
Clinical Manifestations
Neonatal HSV can appear as disseminated multiorgan disease (approximately 25% of neonates with HSV infection); localized disease of the CNS (approximately 35% of neonates); or CNS disease localized to the skin, eyes, and mouth (approximately 40% of neonates) (842). Infants with disseminated disease usually present at age 9--11 days; encephalitis occurs in 60%--75% of these infants. Vesicular rash occurs in approximately 80% of children with localized disease of the skin, eyes, and mouth, but in only approximately 60% of children with CNS or disseminated disease (842,843). Localized disease usually presents by the 10th day of life, and even with treatment, neonates with skin lesions commonly have cutaneous recurrences during the first 6 months after treatment (842). Infants with localized CNS disease, or CNS disease with skin, eyes, and mouth involvement, usually are ill by day 16--19 of life (843). Although treatment has reduced morbidity and mortality, infants with neonatal HSV infection remain at risk for neurologic sequelae, with the most severe neurologic sequelae seen in those with CNS disease. Two percent to 6% of infants with localized skin, eye, or mucus membrane disease have later neurologic sequelae after apparently successful treatment (844,845).
After the neonatal period, the most common appearance of HSV infection in children is orolabial disease. Fever; irritability; tender submandibular lymphadenopathy; and superficial, painful ulcers in the gingival and oral mucosa and perioral area characterize primary HSV gingivostomatitis. HIV-infected children who experience primary infection when they are immunocompromised can have severe local lesions or, more rarely, disseminated HSV with visceral involvement and generalized skin lesions with primary infection. Other sites of involvement among HIV-infected children with severe immunocompromise include the esophagus, CNS, and genitals and disseminated disease involving the liver, adrenals, lung, kidney, spleen, and brain.
HSV genitalis is the more common manifestation of HSV-2 infection. Painful, ulcerative lesions on the perineum and on vaginal and urethral mucosal surfaces are common during primary infection. Local symptoms include pain and pruritis. Mucosal disease usually is accompanied by dysuria, or vaginal or urethral discharge; inguinal lymphadenopathy, particularly in primary infection, is common with perineal disease (846).
Among HIV-infected persons, HSV keratitis, neonatal HSV, HSV encephalitis, and herpetic whitlow are similar in presentation and treatment to diseases in HIV-uninfected persons but might be more severe. HSV retinitis occurs as acute retinal necrosis, occasionally in patients with HSV encephalitis. HSV encephalitis occurs among HIV-infected persons, but no evidence indicates it is more severe or common than among HIV-uninfected persons.
Diagnosis
Clinical diagnosis is based on the typical appearance of vesicles and ulcers. The virus can be isolated in culture and usually can be detected in tissue culture cells within 1--3 days. HSV DNA by PCR also can be used to establish the presence of HSV infection in skin lesions or infected mucosal sites of perinatally exposed newborns. To diagnose neonatal HSV infection, specimens for HSV culture or HSV DNA PCR should be obtained from blood and skin vesicles, mouth or nasopharynx, eyes, urine, and stool or rectum; positive cultures from any of the latter sites >48 hours after birth indicate viral replication rather than contamination after intrapartum exposure. CSF should be tested for HSV DNA by PCR amplification of an HSV DNA sequence common to both HSV-1 and HSV-2.
Direct immunofluorescence for HSV antigen can be conducted on cells collected from skin, conjunctiva, or mucosal lesion scrapings. Giemsa staining (Tzanck preparation) of lesion cell scrapings might show multinucleated giant cells and eosinophilic intranuclear inclusions, but this test is insensitive, nonspecific, and not routinely recommended.
Among children with suspected HSV encephalitis, cultures of the CSF for HSV usually are negative. Detection of HSV DNA by PCR in the CSF has replaced brain biopsy as the diagnostic test of choice for such patients. The reported sensitivity of the CSF PCR in neonatal CNS HSV disease is 75%--100%, with the specificity 70%--100% (847). During therapy for HSV-proven encephalitis, the CSF HSV PCR remains positive for a mean of 10 days (848).
Definitive diagnosis of HSV esophagitis requires endoscopy with biopsy (histologic evidence of multinucleated giant cells with intranuclear viral inclusion) and culture.
Prevention Recommendations
Preventing Exposure
The rate of HSV transmission to the fetus and neonate among HIV-infected pregnant women coinfected with HSV is not known. Although isolated cases of in utero HSV transmission with primary HSV infection during pregnancy among HIV-uninfected women have been reported, the predominant risk, regardless of HIV coinfection, is from maternal genital shedding at delivery. Effective HAART regimens may decrease, but not prevent, maternal genital HSV shedding and recurrence of genital lesions (849).
Use of acyclovir or valacyclovir near term suppresses genital herpes outbreaks and shedding in late pregnancy among HIV-uninfected women with HSV infection and appears to reduce the need for cesarean delivery for recurrent HSV (850--852). However, the safety and efficacy of this strategy have not been evaluated among HIV-infected women who are more likely to have antibody to HSV-2 and to have both symptomatic and asymptomatic reactivation of genital HSV. Therefore, the use of acyclovir or valacyclovir specifically to reduce the need for cesarean delivery among HIV/HSV-coinfected women is not recommended (DIII) (853). In addition, cases have been reported of HSV-infected neonates born to women who received suppressive antiviral therapy near term (854).
For pregnant women who have active genital HSV at the onset of labor, delivery by elective cesarean delivery, preferably before rupture of membranes, is recommended (AI) (855).
For the HIV-infected child, exposure to HSV-1 is an inevitable part of childhood, and no proven ways exist to prevent exposure. Direct contact of children with secretions from active HSV lesions, such as herpes labialis, on the mother, household member, or other persons should be avoided.
Among sexually active HIV-infected adults, latex condoms should be used during every act of sexual intercourse to reduce the risk for exposure to HSV and other sexually transmitted pathogens (AII). They should specifically avoid sexual contact when herpetic lesions (genital or orolabial) are evident (AII). Data suggest that chronic suppressive therapy with valacyclovir in persons with genital herpes reduced HSV-2 transmission to susceptible heterosexual partners by 50%. HIV-infected adults receiving HAART had fewer symptomatic herpetic lesions than did HIV-infected adults not receiving HAART, but mucosal HSV-2 shedding was similar (856).
Preventing First Episode of Disease
Antiviral prophylaxis after exposure to HSV, or to prevent initial episodes of HSV disease among persons with latent infection, is not recommended (DIII).
Treatment Recommendations
Treatment of Disease
Acyclovir is the drug of choice for treatment of local and disseminated HSV among infants and children, regardless of HIV-infection status (AI). Both oral and IV preparations are available. Neonatal HSV disease should be treated with high-dose IV acyclovir (20 mg/kg/dose three times daily) administered for 21 days for CNS and disseminated disease, and for 14 days for disease of the skin, eyes, and mouth (AI) (857). Acyclovir therapy should not be discontinued in neonates with CNS disease unless a repeat CSF HSV DNA PCR assay is negative near the end of treatment (BIII). Orolabial lesions in HIV-infected children can be treated with oral acyclovir for 5--10 days (AI). Moderate-to-severe mucocutaneous HSV lesions are best treated initially with IV acyclovir (AI). Patients may be switched to oral therapy after the lesions have begun to regress, and therapy continued until lesions have completely healed. Acyclovir is the drug of choice for disseminated HSV and HSV encephalitis in children. Regardless of patient age, HSV encephalitis should be treated for 21 days (AII). Genital HSV should be treated with oral acyclovir for 5--14 days (AI). Trifluridine, a fluorinated pyrimidine nucleoside, is the treatment of choice for herpes keratoconjunctivitis, one drop onto the cornea (858) every 2 hours, not to exceed nine drops/day; it is not recommended for longer than 21 days (AII).
Alternatives to acyclovir in older adolescents and adults include valacyclovir, famciclovir, and foscarnet (AI) (859-862). Valacyclovir is a prodrug of acyclovir with improved bioavailability that is rapidly converted to acyclovir after absorption. Data are limited on valacyclovir use in children (863); bioavailability is approximately 45% and independent of age in children. Limited data indicate pediatric blood levels of acyclovir (from the prodrug valacyclovir) similar to levels achieved with valacyclovir tablets in adults can be achieved by an oral dose of valacyclovir of 20−25 mg/kg/dose administered two to three times a day (864). However, no pediatric formulation is available; thus, this drug is an alternative only for children old enough to swallow the large valacyclovir tablets. The tablets can be crushed, but they have an unpleasant taste. No specific data exist on the pharmacokinetics and dosing of famciclovir in children, and no pediatric preparation is available (864). Intravenous foscarnet may be used in cases of acyclovir-resistant herpes infection (BIII).
Monitoring and Adverse Events, Including IRIS
Acyclovir is excreted primarily by the kidney; as a result, dose adjustment based on creatinine clearance is needed in patients with renal insufficiency or renal failure. Primary toxicities of acyclovir are phlebitis, renal toxicity, nausea, vomiting, and rash. Toxicities are similar for valacyclovir. In infants receiving high-dose acyclovir for neonatal disease, the major side effect was neutropenia (e.g., absolute neutrophil count <1000/mm3) (857). Grade 3 or higher nephrotoxicity was observed in 6%. For infants and children receiving high-dose IV acyclovir, monitoring of complete blood counts and renal function is recommended at initiation of treatment and once or twice weekly for the duration of treatment, particularly for those with underlying renal dysfunction or those receiving prolonged therapy.
Management of acyclovir-resistant herpes with foscarnet is associated with decreased renal function; ≤30% of patients experience increases in serum creatinine levels. Renal toxicity and foscarnet binding to divalent metal ions such as calcium leads to metabolic abnormalities in approximately one third of patients, and serious electrolyte imbalances (including abnormalities in calcium, phosphorus, magnesium, and potassium levels) and secondary seizures, or cardiac dysrhythmias can occur. Abnormal liver transaminases and CNS symptoms also can occur. For patients receiving foscarnet, complete blood counts, serum electrolytes, and renal function should be monitored twice weekly during induction therapy and once weekly thereafter.
Atypical lesions that may have a delayed response to therapy have been reported in adults initiating HAART and attributed to IRIS (856).
Management of Treatment Failure
Treatment failure related to resistance to antiviral drugs should be suspected if lesions do not indicate signs of resolution within 7--10 days after initiation of therapy. In immunocompromised patients with suspected acyclovir-resistant HSV, a lesion culture should be obtained and, if virus is isolated, susceptibility testing performed to confirm drug resistance (858).
The treatment of choice for acyclovir-resistant HSV is IV foscarnet (AI) (858,865). All acyclovir-resistant HSV strains are resistant to valacyclovir, and most are resistant to famciclovir. Topical trifluridine or cidofovir also have been used successfully for lesions on cutaneous surfaces, although prolonged application for 21--28 days or longer might be required (866). IV cidofovir has been used to treat a child with acyclovir and foscarnet resistant HSV (867).
Prevention of Recurrence
Administration of oral acyclovir prevented cutaneous recurrences of HSV after neonatal disease of the skin, eyes, and mouth. However, the effect of such therapy on neurologic outcome needs assessment, and additional investigation is necessary before routine use of suppressive therapy can be recommended for children (845).
Because episodes of HSV disease can be treated successfully, chronic therapy with acyclovir is not required after lesions resolve. However, children who have frequent or severe recurrences (e.g., four to six severe episodes a year) can be administered daily suppressive therapy with oral acyclovir (AI) (864). Valacyclovir or famciclovir also are options for older children (AI). Effective HAART may decrease recurrences.
Discontinuing Secondary Prophylaxis
Not applicable, secondary prophylaxis not usually recommended in children.
Human Papillomavirus
Epidemiology
Human papillomavirus (HPV) infects cutaneous and mucosal squamous epithelium. More than 100 distinct types of HPV exist (868). They can be categorized on the basis of the site at which they occur (genital, cutaneous) and as high- or low-risk on the basis of their potential to induce malignant proliferation. Of the approximately 40 genital (i.e., muscosal) HPV types, 12 types are identified as established high risk (HPV 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59), and six, as probable high risk (HPV 26, 53, 66, 68, 73, 82) (868). Genital HPV types can infect the genitals, conjunctiva, mouth, throat, and respiratory tract. Nongenital (i.e., cutaneous) HPV types that cause skin warts (such as HPV 2) are distinct from those causing genital infections. Children with compromised cellular immunity might have intense and widespread appearance of both cutaneous and mucosal warts.
HPV-associated cutaneous warts are transmitted by close person-to-person contact that is facilitated by minor trauma to the skin. HPV-associated anogenital warts are transmitted by sexual contact but also might be acquired at delivery. There is some evidence that transmission may occur from nongenital sites. Genital warts (condyloma accuminatum) in young children might be a sign of sexual abuse (869,870).
Mother-to-child transmission of HPV is not surprising because pregnant women have high rates of HPV infection (871,872). HPV DNA has been identified in 5%--42% of pregnant women without HIV infection (873--875). Although a few studies have demonstrated an increased prevalence of detectable HPV DNA during pregnancy, this finding has not been consistent across studies (874,875). Among nonpregnant women, HPV DNA is detected more frequently among HIV-infected than HIV-uninfected women, with reported prevalence rates of 12%--77% (876,877). Few data have been reported about HPV prevalence in HIV-infected pregnant women, with one study reporting a prevalence of 35% (878).
HPV DNA has been detected in cord blood and amniotic fluid, indicating the potential for in utero infection (879,880). Duration of membrane rupture has been associated with mother-to-child HPV transmission, and reports of HPV transmission to infants born by cesarean delivery suggest that HPV can cross the placental barrier (881,882). Most transmission studies report on the detection of HPV DNA because clinical disease (i.e., warts) is rare in infants. Reported rates of HPV DNA detection in nasopharyngeal aspirates, buccal brush swabs, or genital swabs from infants born to HPV-infected mothers have varied from 2% to 80% (871,872,881). Neonatal clinical abnormalities at birth are rare. Genital condylomata can occur within weeks to months after birth but are rare. Respiratory papillomatosis, a rare condition in which respiratory papillomas develop and typically recur (i.e., juvenile onset recurrent respiratory papillomatosis), usually manifests within 2--5 years after birth. Respiratory papillomatosis in children is thought to be secondary to HPV transmitted from mother to child through aspiration of infectious maternal genital secretions during delivery; however, other methods of transmission are possible (871). A recent parent-child study found that the cumulative detection rate for high-risk HPV from the child's genital samples was 53% over 3 years; 1.5% persisted with the same HPV type at the end of study. During the same period, HPV was detected more commonly from oral samples, with a cumulative rate of 63% with 10% persistence (871). Persistence in infant oral samples was associated with persistent oral HPV detection in the mothers. Persistent genital infections were associated with the mother's history of genital warts. The prevalence of HPV DNA in the oropharynx in 1235 children in Iowa was assessed in one study; a bimodal age distribution was found, with highest HPV prevalence in the youngest and oldest groups: 2.5% at age <1 year, 0.8% at 1--4 years, 1.2% at 5--11 years, 1.5% at 12--15 years, and 3.3% at 16--20 years (883). These data support the high rate of HPV clearance in children, even if the infant is exposed to HPV during infancy.
Although perinatal transmission is possible, genital HPV is most commonly sexually transmitted. Young age at first intercourse and a higher number of recent sex partners are strong risk factors for HPV (884--888). Prevalence of HPV is common in sexually active adolescent girls, with prevalences of 12%--64% compared with 2%--7% in women aged >35 years (885,889--891). Cervical HPV is acquired shortly after onset of sexual activity, with 50% cumulative exposure within 3 years (884,886), even among young women with one sex partner (892).
Although the incidence of anogenital HPV infection in sexually active youth is high, longitudinal studies have demonstrated that 80%--90% of infections among youth without HIV infection might be transient and spontaneously regress (890,893). Repeated infections with new types are common (889), but whether repeat detection of same HPV type infections result from new exposures or from reactivation of latent infection is not known (894). Even though the prevalence of HPV consistently has been higher among HIV-infected men and women, acquisition rates of HPV appear similar among HIV-infected and HIV-uninfected persons (895). The higher prevalence results from the increased rate of HPV persistence in HIV-infected persons. In one study of adolescents with HIV, only 50% cleared their HPV infections (895). Other possible risk factors for persistence have included multiple types or high-risk types of HPV (e.g., 16 and 18), older age, smoking cigarettes, and duration of HPV detection for >12 months (896). Detection of anal HPV also is higher among HIV-infected youth (897). Receptive anal sex is a risk for anal HPV among HIV-infected and HIV-uninfected men; the association between anal HPV infection and anal sex is not as clear for women (897,898). In one study of HIV-infected women, anal HPV infection was more prevalent than cervical infection (899).
Persistent infection with high-risk HPV is associated with increased risk for cervical intraepithelial neoplasia (CIN) and cervical and vulvovaginal carcinoma in women and for anal intraepithelial neoplasia and anal carcinoma in both women and men. Rates of all these HPV-associated cancers are higher among HIV-infected persons (900) and believed to result predominantly from the increased risk for persistent infection in this group. Interestingly, the risk for these HPV-associated cancers is highest among young persons with HIV (900). Adolescent girls differ biologically from adult women (e.g., cervical squamous metaplasia) that might increase their susceptibility to either persistent infection or disease (877,901). The risk for HPV-associated cervical abnormalities increases among HIV-infected youth. In one study, 33% of HPV-infected youth with HIV progressed to high-grade squamous intraepithelial lesions (HSIL) within 3 years of observation (902). CD4 immunosuppression correlated with HPV persistence but not with HSIL. Even though HAART has dramatically altered HIV's natural history, its impact on HPV and HPV-associated neoplasia is less clear. Other risks associated with cervical cancers include lack of cervical cancer screening, prolonged hormonal contraceptive use, parity, smoking, and immunocompromising conditions (other than HIV) (888).
Clinical Manifestations
Genital HPV types cause hyperplastic, papillomatous, and verrucous squamous epithelial lesions on skin and mucus membranes, including anal, genital, oral, nasal, conjunctival, GI, bladder, and respiratory tract mucosa. Wart lesions can appear as papules, flat, smooth, or pedunculated lesions. Sometimes they can be soft, pink, or white "cauliflower-like" sessile growths on moist mucosal surfaces (condyloma accuminatum), or keratotic lesions on squamous epithelium of the skin with a thick, horny layer. Because HPV requires access to basal epithelial cells through disruption of the squamous epithelium, common sites for skin warts are the hand, elbows, knees, and feet.
Diagnosis
Most cutaneous and anogenital warts can be diagnosed by physical examination. Diagnosis of laryngeal papillomas requires laryngoscopy, and children with suspected respiratory tract papillomas need to be evaluated by a pediatric otolaryngologist.
Whether all cervical HPV infections result in microscopic abnormalities described as squamous intra-epithelial lesions (SIL) remains debatable. Most HPV DNA detected from cervical samples is associated with normal cervical cytology. These infections either have microscopic lesions not detected by the insensitive cytologic test or are truly latent. The Pap test (conventional or liquid-based) is a screening test for HPV-associated disease, including cervical cancer. However, histology remains the gold standard for confirming HPV-associated cervical and anal precancerous lesions and invasive cancers.
Biopsies for histologic diagnosis usually are directed by colposcopy or high-resolution anoscopy. No screening tests are available for vaginal and vulvar disease; however, these lesions often are diagnosed in women referred to colposcopy for abnormal cytology or because of abnormalities noted on macroscopic examination. Histology also should be confirmed for vulvar and vaginal squamous intraepithelial lesions and cancer.
HPV DNA can be detected using several platforms (903). The only FDA-approved test is HybridCapture, which detects any of 13 high-risk HPV types and is not type specific. Detection and typing of HPV is not recommended for diagnosing or managing anogenital or cutaneous warts or papillomas (870). HPV testing is not recommended in any circumstance for adolescent girls (aged ≤20 years) (904), regardless of whether they are HIV-infected or HIV-uninfected, because of the high rates of HPV infection. These recommendations may change once specific HPV testing has been studied in clinical trials.
Prevention Recommendations
Preventing Exposure
HIV-infected persons should use latex condoms during every act of sexual intercourse to reduce the risk for exposure to sexually transmitted pathogens (AII), including HPV (887).
HPV vaccine
In June 2006, FDA approved the first preventive vaccine for HPV types 16, 18, 6, and 11. HPV 16 and 18 cause almost 70% of invasive cervical cancers, and HPV 6 and 11 cause 90% of external genital warts. HPV exposure is extremely common after sexual contact, not just sexual intercourse, is initiated. Administration of the vaccine is critical before onset of sexual activity for it to be fully effective. Data for HIV-uninfected women showed efficacy rates of 95% for preventing HPV infection and high-grade CIN related to vaccine-related HPV strains and 99% efficacy for genital warts (905,906). However, if previous exposure to the vaccine HPV types was documented, no efficacy was noted for that type, underscoring the fact that the vaccine is not therapeutic. A second vaccine targeting HPV 16 and 18 has had similar efficacy (Cervarix, Glaxo Smith Kline, Research Triangle Park, North Carolina) (907) and is expected to receive FDA approval in 2009.
Although the vaccines are considered safe, studies in HIV-infected persons are not yet available, so immunogenicity and efficacy in this population has not been established. However, because quadrivalent HPV vaccine is noninfectious, it can be administered to females who are immunosuppressed by disease (including HIV) or medications (AI); immune response and vaccine efficacy might be less than in immunocompetent persons (Figure 2) (30,908). Studies of the immunogenicity and safety of the HPV vaccine in HIV-infected children show that the vaccine is safe. Although seroconversion occurred in 100% of those HIV-infected seronegative children, titers for HPV 6 and 18 were lower than those for healthy children (909). CDC recommendations for HPV vaccination of all children and adolescents should be followed for both HIV-infected and HIV-uninfected persons (908). The first dose of the HPV vaccine series should be administered to girls aged 11--12 years but can be administered as early as 9 years. The second dose should be administered 2 months after the first dose, and the third dose should be administered 6 months after the first dose. HIV-infected adolescent girls and young women aged 13--26 years who have not been previously vaccinated also should be vaccinated with the three-dose HPV vaccine series.
The HPV vaccine does not have any therapeutic benefit for existing HPV-related lesions in either HIV-infected or HIV-uninfected women. No studies using the HPV vaccine to prevent HPV infection and associated lesions of the anus, penis, or oral cavity in men have been published, and the vaccine is not approved for use in men in the United States. As in HIV-infected women, no data exist on the safety or efficacy of the HPV vaccine in HIV-infected men.
Preventing First Episode of Disease
HPV-associated genital epithelial cancers among HIV-infected women
As part of a complete history and physical examination, HIV-infected sexually active women should have a pelvic examination and a cervical cancer screening test (Pap test, either conventional or liquid-based). In accordance with the recommendation of the Agency for Health Care Policy and Research, a Pap smear should be obtained twice during the first year after diagnosis of HIV infection and, if the results are normal, annually thereafter (AII). If the results of the Pap smear are abnormal, care should be provided according to treatment guidelines described below for adolescents. Adult women (e.g., aged >20 years) should be managed according to adult guidelines. No data are available that demonstrate these guidelines should be modified to prevent cervical disease for women on HAART.
HPV-associated anal intraepithelial neoplasia and anal cancer among HIV-infected women and HIV-infected men who have sex with men
Evidence from multiple studies demonstrates that HIV-infected men who have sex with men and HIV-infected women are at increased risk for anal HSIL and might be at increased risk for anal cancer. In view of this evidence, and given a cost-effectiveness analysis projecting that screening and treatment for anal HSILs provide clinical benefits comparable to other measures to prevent OIs among HIV-infected persons (910), anal cytology screening of HIV-infected men who have sex with men and of women might become a useful preventive measure (911). However, studies of screening and treatment programs for anal HSIL need to be implemented before recommendations for anal cytology screening can be made.
Treatment Recommendations
Treatment of Disease
Genital warts
Multiple treatments for HPV-associated skin and external genital lesions exist, but no one treatment is ideal for all patients or all lesions (CIII) (870). Standard topical therapy for HPV-associated lesions among HIV-infected children often is ineffective. Treatment can induce wart-free periods, but the underlying viral infection can persist and result in recurrence. No data suggest that treatment modalities for external genital warts should differ in HIV-infected persons. However, persons who are immunosuppressed because of HIV might have larger or more numerous warts, might not respond as well as immunocompetent persons to therapy for genital warts, and might have more frequent recurrences after treatment (154,912,913). In addition, topical treatments are seldom effective in patients with large or extensive lesions. Topical treatments include podofilox (0.5%) solution or gel (antimitotic agent), imiquimod (5%) cream (topical immune enhancer that stimulates production of interferon and other cytokines), trichloroacetic or bichloroacetic acid (80%--90% aqueous solution) (caustic agents that destroy warts by chemical coagulation of proteins), and podophyllin resin (10%--25%) in a compound tincture of benzoin (contains antimitotic compounds and mutagens). Podofilox and imiquimod are patient-applied. Podofilox is applied to all lesions twice a day for 3 consecutive days, followed by 4 days of no therapy. This cycle can be repeated weekly up to 4 weeks (BIII). Imiquimod is applied once daily at bedtime for 3 nonconsecutive nights a week for up to 16 weeks. The treatment area should be washed with soap and water the following morning (BII). Acid cauterization (i.e., tricholoracetic or bichloroacetic acid) and podophyllin resin require application by a health-care provider. Acid cauterization should be discontinued if substantial improvement is not observed after three treatment sessions or complete clearance has not occurred after six consecutive treatments (BIII). Podophyllin resin is applied and removed by washing a few hours later; applications can be repeated weekly for up to 6 weeks (CIII). Podophyllin resin has lost favor because the resin can vary in potency and is not reliable.
Other treatments include Veregen(r) (based on the antioxidative effect of green tea extract), intralesion interferon or 5-fluorouracil/epinephrinegel implant, and cidofovir topical gel (1%). Veregen (sinecatechins) is a new FDA-approved topical product for external genital wart treatment that can be used three times daily for up to 16 weeks. No data are available on this treatment for HIV-infected persons (CIII). Cidofovir topical gel (1%) is a topical preparation that has been evaluated in a limited number of adults for treatment of anogenital HPV infection (CIII). Topical cidofovir can be absorbed systemically and be associated with renal toxicity (914). Injectable therapy (e.g., interferon or 5-fluoracil/epinephringel implant) should be offered in only severe recalcitrant cases because of inconvenient routes of administration, frequent office visits, and a high frequency of systemic adverse effects.
Lesions can be removed by cryotherapy or surgery (BIII). Cryotherapy (application of liquid nitrogen or dry ice) must be applied until each lesion is thoroughly frozen. Treatment can be repeated every 1--2 weeks up to four times. The major toxicity is local pain. Adequate local pain management is essential for all caustic treatments. Topical anesthetics are favored. Lesions can be removed surgically by tangential scissor, tangential shave excision, curettage, or electrosurgery.
Oral warts
Oral warts can be located on various surfaces in the mouth. In contrast to other oral manifestations of HIV, an increased prevalence of oral warts in patients on HAART has been reported from the United States and the United Kingdom (915,916). No randomized trials have been conducted on treatment of oral warts. Treatments include surgical excision and cryotherapy. Some topical modalities have had success (917).
Respiratory papillomatosis
Respiratory papillomatosis should be managed by a specialist (918). Treatment is directed toward removing lesions obstructing the airway rather than at the elimination of disease. Lesions are removed by debridement or laser. Systemic interferon-alfa therapy or intralesional cidofovir has been used as an investigational treatment with limited success in children, with frequent recurrences or extension into the trachea, bronchi, or lung parenchyma (CIII).
Management of abnormal cytology
Management of anogenital HPV infection accompanied by cytologic changes indicating dysplasia/carcinoma among adolescents differs slightly from that for adults. Adolescents aged 13 --20 years and young women are considered a special population. The risk for invasive cervical cancer is low in this group, but CIN lesions are common. As noted earlier, CIN in HIV-uninfected adolescents also has a high rate of spontaneous regression of CIN lesions (919). HPV testing for follow-up is not recommended for adolescents, regardless of HIV infection status (904).
Because of the high rate of progression to HSIL, all HIV-infected adolescents with any SIL, either low-grade (LSIL) or high-grade (HSIL), or atypical squamous cells of undetermined significance (ASCUS) suggestive of HSIL should be referred for colposcopy (BIII). In patients with ASCUS alone, Pap smear for cytology can be repeated in 6--12 months. If ASCUS or greater is found on repeat cytology, referral to colposcopy is warranted.
Treatment of histologic CIN
Follow-up with annual cytologic assessment is recommended for adolescents with CIN 1 (AII) (904). At the 12-month follow-up, only adolescents with HSIL or greater on the repeat cytology should be referred back to colposcopy. At the 24-month follow-up, those with an ASCUS or greater result should be referred back to colposcopy (AII).
For adolescent girls and young women with a histologic diagnosis of CIN 2 or 3 not otherwise specified, either treatment or observation for up to 24 months using both colposcopy and cytology at 6-month intervals is acceptable, provided colposcopy is satisfactory (BIII) (904). When a histologic diagnosis of CIN 2 is specified, observation is preferred, but treatment is acceptable. When CIN 3 is histologically diagnosed or when colposcopy is unsatisfactory, treatment is recommended (BIII).
If the colposcopic appearance of the lesion worsens or if HSIL cytology or a high-grade colposcopic lesion persists for 1 year, repeat biopsy is recommended (BIII). After two consecutive "negative for intraepithelial lesion or malignancy" results, adolescents and young women with normal colposcopy can return to routine cytologic screening (BII). Treatment is recommended if CIN 3 is subsequently identified or if CIN 2 or 3 persists for 24 months (BII).
Persistent CIN 1, 2, and 3 lesions in HIV-infected women should be treated as in HIV-uninfected women (904). Conventional therapies used to treat CIN 2 or 3 include cryotherapy, laser therapy, cone biopsy, and a loop electrosurgical excision procedure (LEEP). LEEP usually is the preferred mode of treatment (BIII). Recurrence rates of 40%--60% after treatment have been reported among HIV-infected women undergoing these procedures. Pregnant HIV-infected adolescents should be treated similarly to pregnant HIV-infected women.
Role of antiretroviral therapy
HAART has not been consistently associated with reduced risk for HPV-related cervical abnormalities in HIV-infected women. However, severe immunosuppression is associated with higher morbidity and mortality.
Monitoring of Adverse Events, Including IRIS
Monitoring is required during and after treatment of genital warts because each treatment has associated toxicity and recurrences are common after treatment. Patients can be monitored by physical examination for evidence of recurrence. The major toxicity of podophyllotoxin and topical podophyllin resin is local skin irritation. Also, if podophyllin is applied to a large treatment area, systemic absorption can cause nausea, vomiting, and CNS effects. The major toxicity of imiquimod is inflammation at the application site. The major toxicity of cryotherapy is local pain. The major side effects of surgical treatment for genital warts are local pain, bleeding, and secondary infection. The major adverse events associated with acid cauterization are local pain and irritation or ulceration of adjacent normal skin. Intralesional interferon can be associated with systemic toxicities of interferon, including fever, fatigue, myalgia, malaise, depression, and other influenza-like symptoms. Infrared coagulation may lead to bleeding and abscess formation. Scarring can occur with any of the above treatment modalities. Topical cidofovir may result in systemic absorption and be associated with renal toxicity (914). Secondary infections are not uncommon if ulcerations occur. Patients should be monitored regularly after each treatment.
Because risk for recurrence of CIN and cervical cancer after conventional therapy increases among HIV-seropositive persons, patients should be carefully followed after treatment with frequent cytologic screening and colposcopic examination according to published guidelines (AII) (920,921). Treatment of CIN with ablative and excisional modalities can be associated with several adverse events such as pain and discomfort, intraoperative hemorrhage, postoperative hemorrhage, infection and cervical stenosis.
The major toxicity of topical agents for treatment of external genital warts is local pain or irritation of adjacent normal skin. HIV-infected patients with immunosuppression might have a lower response rate to all of these modalities. Secondary infections are not uncommon if ulcerations occur. Patients should be monitored regularly after each treatment.
Because of the frequent recurrence of squamous intraepithelial lesions after treatment, close surveillance with colposcopy and cytology are recommended.
An "immune reconstitution"-like syndrome related to HPV-associated oral warts among HIV-infected adults has been observed in which the occurrence of oral warts was associated with decreased HIV RNA levels with HAART (915). Immune reconstitution in response to viral load reduction might result in a return of marked inflammatory responses against latent oral HPV infection.
Management of Treatment Failure
Treatment failure is defined as the persistence or recurrence of lesions after appropriate therapy. For persistent or recurrent genital warts, retreatment with any of the modalities previously described should be considered, preferably with an alternative modality to the one that previously failed (AIII). Genital warts often require more than one course of treatment. Recalcitrant warts should be managed by experienced clinicians and referred for excisional therapy. Recurrence of CIN may require additional treatments (i.e., LEEP and laser).
Prevention of Recurrence
No recommendations exist for preventing recurrence of external genital warts. Patients should be monitored with cytologic screening according to published guidelines and, when indicated, colposcopic examination for recurrent lesions (AI) (922,923). In one study, use of low-dose intravaginal flurouracil reduced recurrence of CIN after LEEP, but lack of additional studies do not warrant routine use (924). Efudex should not be used in pregnant women.
Discontinuing Secondary Prophylaxis
Not applicable.
Progressive Multifocal Leukodystrophy
Epidemiology
First described in association with chronic lymphocytic leukemia and Hodgkin disease, progressive multifocal leukoencephalopathy (PML) is a rare demyelinating disease of the CNS that occurs in immunocompromised patients (925). It is caused by primary infection or reactivation of Jamestown Canyon virus (JCV), a ubiquitous polyoma virus. Most humans are infected with JCV early in life; 50% of children are seropositive by age 9--11 years, and seropositivity among adult women aged >25 years is 72% (926). Infection results in chronic asymptomatic carriage in the kidneys, lymphoid tissue, bone marrow, and lymphocytes (927,928); in a patient with a weakened immune system, the virus may reactivate and spread to the brain by lymphocytes causing neurologic dysfunction and serious and life-threatening disease.
PML is an AIDS-defining illness in HIV-infected persons. PML has been rare in reports from large series of HIV-infected children (1,4,929), but cases have been reported in children (930--935).
Clinical Manifestations
No symptoms are known to be associated with acute JCV infection. PML is the only disease caused by JCV. Cases in children are similar to those in adults.
Demyelination is at first patchy, involving subcortical regions, and then spreads to deep white matter in confluent pattern; thus, PML initially may present with focal neurologic deficits that can involve different brain regions. The established criteria for clinical diagnosis are focal signs and symptoms on neurologic examination, focal white matter lesions on MRI or CT without mass effect, and exclusion of other causes of the clinical and neuroradiologic findings (936). The disease has an insidious onset and produces a neurologic syndrome that steadily progresses over weeks or months, characterized by confusion, disorientation, lack of energy, loss of balance, cognitive dysfunction, dementia, seizures, ataxia, aphasia, cranial nerve deficits, visual abnormalities (e.g., blurred or double vision or loss of vision), hemiparesis or quadraparesis, and eventually coma. During the pre-HAART era, adults and children with PML had extremely poor survival (929). Survival among adults has improved during the HAART era (937--939). No comparable data exist for children.
Diagnosis
A confirmed diagnosis of PML requires a compatible clinical syndrome and radiographic findings coupled with brain biopsy demonstrating a characteristic triad of pathologic foci of demyelination, enlarged hyperchromatic oligodendrocytes with enlarged nuclei and basophilic-staining intranuclear material, and enlarged astrocytes with bizarre hyperchromatic nuclei. When only two of these features are present, JCV can be demonstrated by in situ hybridization or by electron microscopy for definitive diagnosis.
Although brain biopsy remains the confirmative test for diagnosis of PML, brain scans such as MRI or CT can reveal lesions in the brain. The radiologic features of PML are typically noninflammatory (unless associated with HAART-related IRIS). Typical CT abnormalities include single or multiple hypodense, nonenhancing cerebral white matter lesions; cerebellum and brain stem occasionally are involved. MRI depicts white matter lesions of low T1 signal intensity and high proton density on T2 weighted images with absence of edema or mass effects, as might be seen with cerebral toxoplasmosis or lymphoma. Postcontrast enhancement is unusual and, when present, usually is sparse, with a thin or reticulated appearance adjacent to the edge of the lesions.
PML diagnosis is now facilitated by use of PCR to detect JCV DNA in CSF, which may obviate the need for brain biopsy in patients with a compatible clinical syndrome and radiographic findings. Nested JCV PCR on CSF is highly sensitive (90%--100%) and specific (92%--100%) for PML (940). Measurement of JCV DNA levels in CSF samples can be a useful virologic marker for managing PML in patients receiving HAART (941).
Prevention Recommendations
Preventing Exposure
There are no known means of preventing exposure to JCV.
Preventing First Episode of Disease
There are no means of preventing PML in severely immune-suppressed persons. Use of HAART can prevent or reverse severe immunosuppression.
Discontinuing Primary Prophylaxis
No means of primary prophylaxis of JCV infection or development of PML have been demonstrated.
Treatment Recommendations
Treatment of Disease
No effective therapy has been established for JCV infection or PML. Survival in HIV-infected adults with PML has substantially improved during the post-HAART era, with a median survival increase from 14 to 64 weeks (942). A CD4 count of >100 cells/mm3 at PML diagnosis was associated with improved survival, and the use of HAART postdiagnosis of PML also was strongly associated with improved survival (942). Thus, the main approach to treatment involves maximally optimizing antiretroviral therapy to reverse the immunosuppression that interferes with normal host response to this virus (AII).
A number of agents have been proposed or reported anecdotally as more specific treatments for PML, but none have proven effective after greater scrutiny or more extensive study. In a randomized open-label trial of IV and intrathecal cytosine arabinoside (943) and a nonrandomized, open-label trial of cidofovir (944), neither drug was effective in producing clinical improvement of PML and neither is routinely recommended (DII). Immunomodulatory approaches, such as interferon-alfa, also have been described in case reports in HIV-infected adults; however, none have been studied in a prospective, controlled clinical trial and, in one analysis, did not provide any benefit beyond that with HAART (945), and thus are also not routinely recommended (DIII).
Monitoring and Adverse Events, Including IRIS
Neurologic stability or improvement and prolonged survival are associated with reduced JCV DNA and appearance of JCV-specific antibody in CSF of HAART-treated PML patients (946).
When antiretroviral therapy is initiated and CD4 counts rise, certain patients will experience neurologic improvement and others might become neurologically stable; however, reports have documented worsening neurologic manifestations after initiation of HAART (939). In certain instances, this worsening is caused by IRIS (939,947--949), examples of which have occurred in children (21). Other cases may represent the natural history of PML. The underlying etiology and trigger of HAART-associated PML is controversial. One hypothesis postulates a reduction in inhibitory cytokines (e.g., interferon-alfa and interleukin-12) after HAART, thus promoting JCV reactivation within the brain or, by increasing trafficking of JCV-infected peripheral lymphocytes, into the brain (950). JCV infection occurring coincidental to HAART initiation resulting in a beneficial inflammatory response with lack of disease progression is another hypothesis (950), particularly given that JCV infection in children with perinatal HIV infection most often would be acquired during childhood. The overall prevalence of IRIS in children is not known. Inflammatory PML should be suspected in HAART-treated children with advanced HIV disease who show acute neurologic deterioration and contrast-enhancing demyelinating lesions on MRI.
Management of Treatment Failure
PML remission with HAART may take several weeks, and no criteria exist that define progression of disease. A working definition used for HIV-infected adults is continued clinical worsening and continued detection of CSF JCV at 3 months (see Guidelines for the Prevention and Treatment of Opportunistic Infections in HIV-Infected Adults) (16). Some patients' PML worsens despite HAART, either because of IRIS or the natural history of PML. In both cases, HAART should be continued. If HAART fails to suppress HIV RNA or to boost the CD4 count, then attention should focus on modifying and optimizing the antiretroviral treatment (AII). However, in HIV-infected children responding well to HAART but with continued worsening PML, an expert in pediatric HIV infection should be consulted.
Prevention of Recurrence
On the basis of its role in reversing the disease, the main preventive measure is an effective antiretroviral regimen that suppresses HIV viremia and maintains CD4 count (AII).
Discontinuing Secondary Prophylaxis
No means of secondary prophylaxis of JCV infection or PML have been demonstrated.
Varicella-Zoster Virus
Epidemiology
Varicella-zoster virus (VZV) infections occur worldwide. In the prevaccine era, approximately 4 million cases of varicella occurred annually in the United States. Since the institution of universal varicella vaccination for healthy children, the incidence of varicella and its associated morbidity and mortality have decreased by approximately 74%--90% (951). Varicella can cause greater morbidity and mortality in HIV-infected persons than among the general population (952,953).
VZV causes both varicella (a primary infection) and zoster (a secondary infection caused by reactivation of latent VZV acquired during varicella) (954). The incubation period for varicella ranges from 10 to 21 days (average: 14 days). Before the widespread use of varicella vaccine in the United States, the annual rate of varicella infection in children aged <10 years was 9%; by adulthood >95% of persons had antibodies to VZV, indicating a history of primary infection (955).
Once established, VZV latency persists for life. Reactivation, causing clinical zoster, occurs in roughly 25% of people. A decline in specific cellular immunity to VZV contributes significantly to development of zoster (954). HIV-infected persons are at higher risk for zoster, by a factor of 15--25 times, than the general population (956). The incidence of zoster also increased with age, particularly in persons aged >50 years.
VZV is transmitted mainly from skin lesions during illness but also can be transmitted through airborne spread (957). Varicella is highly contagious; clinical infection develops in about 80% of susceptible persons exposed in a household (958). Second attacks of varicella are uncommon (954). Zoster is less contagious than varicella.
Mother-to-child transmission of VZV can occur; however, because most adults are immune, varicella complicating pregnancy is unusual. In one study, 13% of HIV-infected pregnant women lacked immunity to VZV (959). Whether mother-to-child VZV transmission increases among HIV-infected pregnant women who have varicella is unknown. Congenital varicella syndrome occurs in approximately 0.4% (95% CI: 0.05%--1.5%) of infants born to women who have varicella during pregnancy before 13 weeks' gestation and in approximately 2% (95% CI: 0%--5%) of infants born to women who have varicella at 13--20 weeks' gestation (960). This syndrome is not seen among women who develop herpes zoster during pregnancy. Fewer than 100 cases of congenital VZV have been reported, none so far in HIV-infected mothers. However, congenital varicella syndrome may not be recognized. Before the availability of varicella vaccine in the United States, approximately 44 cases occurred each year. Congenital varicella syndrome is characterized by cicatricial skin scarring, limb hypoplasia, and neurologic (e.g., microcephaly, cortical atrophy, seizures, and mental retardation), eye (e.g., chorioretinitis, microphthalmia, and cataracts), renal (e.g., hydroureter, and hydronephrosis) and autonomic nervous system abnormalities (neurogenic bladder, swallowing dysfunction, and aspiration pneumonia) (961--963).
VZV can be transmitted to the fetus in later gestation, resulting in acute neonatal varicella. When the mother develops varicella from 4 days before to 2 days after delivery without passive antibody prophylaxis, the attack rate for infants is approximately 20% and mortality, before the availability of antiviral therapy, was approximately 30% (961). In comparison, if maternal varicella precedes delivery long enough to allow transfer of VZV IgG antibodies across the placenta, infants can develop varicella in the first 5 days of life, but it is rarely severe and usually requires no medical intervention.
Zoster occurs only among persons previously infected with VZV. Zoster used to be common in HIV-infected children who had primary varicella infections when their CD4 counts were normal or mildly suppressed. Before PIs were commonly administered to children, among HIV-infected children with low CD4 percentage (i.e., CD4 <15%) at the time of primary varicella, the rate of subsequent zoster was approximately 70% (952,953). The incidence of zoster among HIV-infected children who developed primary varicella when they were immunocompromised was 467 per 1000 child-years, substantially higher than the rates for immunocompromised HIV-infected adults (98 per 1000 person-years) and for children with leukemia (25 per 1000 child-years). As in adults, the CD4 count in children correlates with the frequency of zoster recurrences (964). The incidence of zoster reportedly increases transiently after institution of PIs (22,965). The cause of this phenomenon is not known; one hypothesis is that it results from return of regulatory T-cell function with transient immune dysregulation. Overall, however, the incidence of herpes zoster in HIV-infected children is lower during the HAART era than it was during the pre-HAART era (3). One reason may be immunization of many HIV-infected children with the Oka vaccine strain. The Oka vaccine strain of VZV is the licensed live attenuated varicella vaccine (marketed by Merck in the United States and by GlaxoSmithKline in other countries).
Clinical Manifestations
Varicella in HIV-infected children may be associated with a prodrome of malaise and fever, followed by pruritic vesiculopapular lesions that are more numerous on the face and trunk than on the extremities. Lesions evolve over 5 days through macular, papular, vesicular, pustular, and crust stages. In profoundly immunocompromised hosts, vesicles can persist for weeks and coalesce to form large lesions resembling a burn. Complications of varicella include superinfection of skin with bacterial pathogens, such as staphylococci and streptococci; neurologic manifestations, such as encephalitis, cerebellar ataxia, and transverse myelitis; and on occasion, vasculitic stroke, hepatitis, and pneumonia.
Initial reports of varicella among HIV-infected children suggested severe disease manifestations (966), but more recent studies support less complicated courses, particularly in children receiving antiretroviral therapy or having higher CD4 counts at the time of infection (952,953,967,968). However, the disease may last longer than normal, and the rate of complications is higher than in otherwise healthy children with varicella (968).
Uncommonly, HIV-infected children can have persistent chronic infection, with continued appearance of new VZV lesions for >1 month after primary or recurrent infection (969). The lesions are characteristically varicelliform at onset but evolve into nonhealing ulcers that become necrotic, crusted, and hyperkeratotic. Persistent lesions may be atypical and lack a vesicular component. Chronic VZV was reported in 14% of HIV-infected children with VZV, usually in children with low CD4 counts (964). The virus may become resistant to acyclovir during prolonged therapy (970).
The classical presentation of zoster is a painful or pruritic unilateral vesicular eruption with a dermatomal distribution. Less typical rashes, however, including those that extend beyond dermatomal boundaries or are bilaterally distributed or generalized, also can represent zoster in HIV-infected children. HIV-infected children can have recurrent episodes of reactivated VZV infection that present with a disseminated rash more similar to varicella than zoster but without visceral dissemination; they also can have multiple episodes of recurrent dermatomal disease (964). Encephalitis without rash resulting from zoster also has been reported (971). Diagnostic laboratory studies are useful if children or adolescents present with unusual clinical manifestations suspected to be caused by zoster. Ruling out HSV infection, which can be confused with VZV skin manifestations, also is important (972).
Retinitis is a complication of VZV infection among HIV-infected patients that occurs in children and adolescents (973). VZV-associated retinitis may be confused with CMV retinitis (974). Progressive outer retinal necrosis is a VZV-associated entity that typically occurs among HIV-infected persons with CD4 counts <50 cells/mm3. This rapidly progressive necrotizing herpetic retinopathy often is associated with dermatomal zoster and is characterized by multifocal retinal opacification with little or no ocular inflammation (975,976) and rapid visual loss. Acute retinal necrosis occurs as a peripheral necrotizing retinitis with yellowish thumbprint lesions, retinal vascular sheathing, and vitritis, with a high rate of visual loss often caused by retinal detachment. This latter syndrome can occur in both immunologically normal and deficient persons. Among patients with HIV infection, acute retinal necrosis can occur at any CD4 count, but more often occurs at higher CD4 counts, and progressive outer retinal necrosis more often occurs at lower CD4 counts.
VZV infection should be suspected in children with unilateral vesicular rashes. Retinitis as a complication of VZV should be suspected when CMV cannot be implicated or with progressive and otherwise unexplained encephalitis and a history of varicella or varicella vaccination.
Diagnosis
Varicella and zoster are diagnosed clinically by the typical appearance of generalized pruritic vesicular rash and fever in the former and a frequently painful or pruritic unilateral vesicular rash in a dermatomal pattern in the latter. Direct immunofluorescence for VZV antigen can be performed on cells collected from skin, conjunctiva, or mucosal lesion scrapings for diagnosis (972). The optimal sensitivity of this method requires obtaining cells from the base of a lesion after unroofing a fresh vesicle. Direct and indirect immunofluorescence or immunoperoxidase methods also can be used to detect VZV-infected cells in tissue sections of lung, liver, brain, or other organs. Giemsa-staining (Tzanck preparation) of scrapings from lesions is nonspecific; detection of multinucleated giant cells does not distinguish between VZV and HSV infection.
VZV can be isolated in cell culture from vesicular fluid or ulcer swabs, but the virus is labile. The specimen must be processed rapidly or kept on dry ice or frozen at -70°C (-94°F) if storage is required for more than a few hours (961). Typical cytopathic effects usually can be detected 5--7 days after inoculation; confirmation by staining with virus-specific antiserum is then needed. Shell vial cultures combine centrifugation and staining with fluorescein-conjugated monoclonal antibodies to detect synthesis of VZV proteins in infected cells. This allows results 1--3 days after inoculation, before cytopathic effect is visible. Standard culture usually is necessary when testing the virus for antiviral susceptibility is anticipated, although PCR also can be used (972,977).
PCR can be used to detect VZV in samples, is extremely sensitive and specific, can differentiate between wild-type and vaccine VZV, and is becoming increasingly available and used. In some laboratories, PCR has replaced culture as the "gold standard" (972). Serologic tests can be used to diagnose VZV infection, noting a substantial increase in antibody titer during convalescence (e.g., 2--3 weeks after illness onset) or VZV IgM antibody. VZV reactivation also can induce VZV-IgM antibodies so their presence does not differentiate primary from recurrent VZV infections (961,972).
Prevention Recommendations
Preventing Exposure
HIV-infected children and adults who have no evidence of immunity to VZV (with no history of varicella or zoster; or who are seronegative for VZV by a sensitive, specific antibody assay; or who lack evidence of age appropriate vaccination) should avoid exposure to persons with varicella or zoster (AII). Household contacts of HIV-infected persons should receive varicella vaccine if they lack evidence of immunity (i.e., have no history of varicella or zoster, are seronegative for HIV, were born in the United States after 1980, or lack evidence of age-appropriate vaccination) so they will be less likely to transmit wild-type VZV to their HIV-infected contacts (AIII) (32).
Preventing Disease
HIV-infected children aged 1--8 years in CDC clinical categories N, A, and B (98) and whose CD4 levels are ≥15% should be considered for vaccination (two doses of monovalent single-antigen varicella vaccine); first dose administered at age 12--15 months and second dose 3 months later (BII) (32). Limited data from a clinical trial in HIV-infected children with these characteristics indicate that the vaccine was well-tolerated and that >80% of subjects had detectable VZV-specific immune response (either antibody or cell immune response or both) at 1 year after vaccination (978,979). Data are not available on safety, immunogenicity, or efficacy of the combination measles-mumps-rubella-varicella vaccine in HIV-infected children, and the combination measles-mumps-rubella-varicella vaccine should not be administered as a substitute for the single-antigen varicella vaccine to HIV-infected children (EIII).
Data are lacking on use of varicella vaccine in older HIV-infected children and adolescents. However, on the basis of expert opinion, the safety of varicella vaccine in HIV-infected persons aged >8 years who have similar levels of immune function (e.g., CD4 count ≥200 cells/mm3) is likely to be similar to that in children aged <8 years. Immunogenicity might be lower in older HIV-infected children, adolescents, and adults. However, weighing the risk for severe disease from wild VZV and potential benefit of vaccination, vaccination (two doses of single-antigen vaccine, administered 3 months apart) for persons with CD4 count ≥200 cells/mm3 in these age groups can be considered (BIII) (32).
HIV-infected children tolerate the vaccine well; as in healthy children, serious vaccine-related adverse events are rare. As with healthy children, vaccinated HIV-infected children who develop mild rashes >2 weeks after vaccination rarely require antiviral therapy for Oka VZV; these rashes usually clear in 3--5 days without treatment. If vaccination of HIV-infected children results in more severe clinical disease, the use of acyclovir to treat the Oka vaccine strain of VZV (which is sensitive to acyclovir) might modify the severity of disease. VZV rashes developing <2 weeks after vaccination, however, usually result from wild-type VZV (32).
HIV-infected children with low CD4 levels (<15%) may develop pneumonia and neurologic manifestations from VZV and should not be vaccinated against varicella (EIII). Vaccination of such children after reconstitution of their immune system (CD4 percentage ≥15%) with antiretroviral therapy, however, can be considered (980). Zoster from the vaccine (Oka strain) has been reported in healthy children and in children with acute lymphocytic leukemia, but it has not been described in HIV-infected children (981,982).
Efficacy studies on preventing varicella in HIV-infected children are not available. In vaccinated healthy children (after one dose) and in children with underlying leukemia (after two doses), the effectiveness of varicella vaccine is about 80%--85% for prevention of clinical infection. The majority of varicella cases in vaccinated children are modified, with fewer lesions (commonly <50), fever, and a shorter duration of illness (32,951).
Because Zostavax is licensed only for use in healthy people aged >60 years to prevent zoster, it should not be given to HIV-infected children. Being immunocompromised is a contraindication for its use. There are no data on safety or efficacy for HIV-infected children (EIII).
For postexposure prophylaxis against varicella, HIV-infected children and adolescents who lack evidence of immunity to VZV (i.e., have no history of varicella or zoster; are seronegative for VZV by a sensitive, specific antibody assay; or lack evidence of age-appropriate vaccination) should be passively immunized as soon as possible and within <96 hours after close contact with a person with varicella or zoster (AIII). Previously this was performed by administering varicella-zoster immune globulin (VZIG), but because licensure of varicella vaccine in the United States resulted in dramatically fewer requests for VZIG, VZIG is no longer produced. A new product, human varicella immune globulin (VariZIG), manufactured in Canada, is the replacement. VariZIG is a lyophilized preparation which, when properly reconstituted, is approximately a 5% solution of IgG that can be administered intramuscularly. VariZIG is available under an investigational new drug application expanded access protocol (available at http://www.fda.gov/cber/infosheets/mphvzig020806.htm) (983). VariZIG can be obtained in the United States, and it has received central institutional review board approval but local institutional review board approval also may be necessary. VariZIG can be obtained 24 hours a day from the sole authorized U.S. distributor (FFF Enterprises, Temecula, California) at 1-800-843-7477 or online at http://www.fffenterprises.com. An alternative to VariZIG for passive immunization is IVIG, 400 mg/kg, administered once. IVIG should be administered within 96 hours after exposure.
Data are lacking about the effectiveness of acyclovir for preventing varicella among susceptible HIV-infected children. Published information about this form of prophylaxis for healthy children is minimal (984--986). If VariZIG is not available or >96 hours have passed since exposure, some experts recommend prophylaxis with acyclovir (80 mg/kg/day, administered four times per day for 5--7 days; beginning day 7--10 after exposure, maximum dose of 80 mg, four times per day) (90). However, the use of acyclovir for prophylaxis in HIV-infected VZV-exposed children has not been studied. For that reason, some experts would consider it prudent to wait until the first appearance of rash to start acyclovir therapy for the VZV-susceptible and VZV-exposed HIV-infected child for whom passive immunization was not administered (CIII).
Treatment Recommendations
Treatment of Disease
On the basis of controlled trials among children with malignancies, acyclovir is the drug of choice for treating VZV infection in HIV-infected children (AI). For varicella, acyclovir should be initiated as soon as possible after initial lesions appear. New lesions can continue to appear for 72 hours after initiation of acyclovir and crusting of all lesions might take 5--7 days. IV acyclovir is recommended for treating primary varicella among HIV-infected children with severe immunosuppression (i.e., CD4 < 15%, CDC Immunologic Category 3) (98) or who have high fever or numerous or deep, necrotic, or hemorrhagic skin lesions (AIII). For children aged <1 year, the dose of acyclovir is 10 mg/kg/dose administered intravenously every 8 hours as a 1-hour infusion. Some health-care providers administer the same dose for children aged ≥1 year, and others base dosage of acyclovir on body surface area among children aged ≥1 year old (500 mg/m2 body surface area/dose intravenously every 8 hours as a 1-hour infusion) (90). Administration is for 7--10 days or until no new lesions appear for 48 hours. Oral administration should be used only to treat primary varicella among HIV-infected children with normal or only slightly decreased CD4 counts (CDC Immunologic Category 1 or 2) (98) and have mild varicella disease (BIII).
Acyclovir is the treatment of choice for zoster among HIV-infected children, administered for 7--10 days, although longer durations of therapy should be considered if lesions are slow to resolve (AII). With zoster, oral acyclovir can be administered because the chance for disseminated, life-threatening disease is less with zoster than with varicella. Initial IV administration should be considered for HIV-infected children with severe immunosuppression (i.e., CD4 <15%, CDC Immunologic Category 3) (98), trigeminal nerve involvement, or extensive multidermatomal zoster (AII). If cutaneous lesions are extensive or if clinical evidence of visceral involvement is observed, IV acyclovir should be initiated and continued until cutaneous lesions and visceral disease are clearly resolving (AII), then change to oral administration can be considered to complete the course of therapy (10--14 days in this situation) (AIII) (987). Doses of acyclovir for treating zoster are the same as those for treating varicella.
Progressive outer retinal necrosis is rapidly progressive and leads to profound loss of vision; prognosis for visual preservation is poor despite aggressive therapy, and optimal therapy is yet to be defined (988,989). Regardless of specific VZV antiviral therapy, optimization of antiretroviral therapy also is recommended. Some experts recommend anti-VZV therapy that includes a combination of IV ganciclovir (5 mg/kg/dose administered intravenously every 12 hours) and foscarnet (90 mg/kg/dose administered intravenously every 12 hours) plus twice weekly intravitreal injections of ganciclovir (2 mg/0.05 mL and/or foscarnet 1.2 mg/0.05 mL) (BIII) (990). In contrast, acute retinal necrosis appears more responsive to antiviral therapy, and one recommended treatment is high-dose IV acyclovir (10--15 mg/kg intravenously every 8 hours for 10--14 days), followed by prolonged (i.e., 4--6 weeks) oral valacyclovir (AIII) (990). Involvement of an ophthalmologist experienced in managing patients with VZV retinitis is strongly recommended (AIII).
Alternatives to acyclovir in older adolescents and adults include valacyclovir and famciclovir. Valacyclovir is a prodrug of acyclovir with improved bioavailability that is rapidly converted to acyclovir after absorption and is approved for treating zoster in adults. It is not active against acyclovir-resistant VZV strains. Data are limited for its use in children (863,991); bioavailability is about 45% and independent of age in children. Limited data indicate that pediatric blood levels of acyclovir (from the prodrug valacyclovir) similar to that of valacyclovir tablets in adults can be achieved by administering an oral dose of valacyclovir of 20--25 mg/kg/dose administered two or three times a day (CIII) (864). However, valacyclovir is available only in a caplet formulation; hence this drug is an alternative only for children old enough to swallow the valacyclovir caplets. Although tablets can be crushed, they have a unpleasant taste. A liquid formulation that is stable for 21 days can be prepared in Ora-Sweet(r) and Syrpalta(r) syrups and stored in amber glass bottles (991).
Famciclovir is the oral prodrug of penciclovir. It is not active against acyclovir-resistant VZV strains. It is comparable in efficacy to oral acyclovir in treatment of immunocompromised adults with localized zoster but has not been approved for this indication. It is available only in tablet form. No specific data exist on the pharmacokinetics and dosing of famciclovir in children, and no pediatric preparation is available (EIII) (864).
Monitoring and Adverse Events, Including IRIS
Acyclovir is excreted primarily by the kidney, and dose adjustment (based on creatinine clearance) is needed for patients with renal insufficiency or renal failure. Primary toxicities of acyclovir are phlebitis, renal toxicity, nausea, vomiting, and rash. Toxicities are similar for valacyclovir. Among infants receiving high-dose acyclovir for neonatal HSV disease, the major toxicity was neutropenia (absolute neutrophil count <1000/mm3), which was observed in 21% of children (857). Grade 3 or higher nephrotoxicity was observed in 6% of children. For children receiving high-dose IV acyclovir, monitoring of renal function is recommended at initiation of treatment, and once or twice weekly for the duration of treatment, particularly for those with underlying renal dysfunction or those receiving prolonged therapy.
In HIV-infected adults, immune reconstitution after initiation of HAART may be associated with increased VZV reactivation (965,992). VZV-associated IRIS after HAART also has been described in HIV-infected children (20,24). In a study of 153 HAART-treated children in Thailand, 19% of children starting HAART experienced IRIS; 22% of IRIS cases were secondary to VZV. In the reported cases, manifestations were typically mild and cutaneous, had a typical dermatomal distribution of vesicular lesions as seen in VZV reactivation, and responded well to treatment with oral acyclovir. Most cases presented in the first 4 months of HAART; the median time from initiation of HAART to onset of clinical symptoms was 6 weeks (range: 2--21 weeks) (24).
Management of Treatment Failure
Children who continue to develop lesions or whose lesions fail to heal after 10 days of treatment may be infected with acyclovir-resistant VZV. If possible, a culture should be obtained to analyze the virus for drug resistance. HIV-infected children with acyclovir-resistant VZV can be treated with IV foscarnet for 7 days or until no new lesions have appeared for 48 hours (AII) (990,993,994). The dose of foscarnet should be administered slowly over the course of 2 hours (i.e., no faster than 1 mg/kg/minute). Infusing foscarnet with saline fluid loading can minimize renal toxicity. Doses should be modified among patients with renal insufficiency.
The main toxicity of foscarnet is decreased renal function; in ≤30% of patients, serum creatinine levels increase. Renal toxicity and foscarnet binding to divalent metal ions (e.g., calcium) leads to metabolic abnormalities in approximately one third of patients. In addition, serious electrolyte imbalances (including abnormalities in calcium, phosphorus, magnesium, and potassium levels), secondary seizures, cardiac dysrhythmias, abnormal liver transaminases, and CNS symptoms can occur.
Prevention of Recurrence
Preventing Recurrence
No preventive measures are available for zoster in HIV-infected children and adolescents. A vaccine to prevent herpes zoster has been approved for use in immunocompetent adults aged >60 years. Data regarding safety and efficacy of this vaccine in HIV-infected persons of any age are lacking, and use of the vaccine in HIV-infected persons is not recommended (DIII). However, prospective clinical trials are planned to evaluate the safety and immunogenicity of herpes zoster vaccine in HIV-infected adults.
Discontinuing Secondary Prophylaxis
Not applicable.
References
- Dankner WM, Lindsey JC, Levin MJ, et al. Correlates of opportunistic infections in children infected with the human immunodeficiency virus managed before highly active antiretroviral therapy. Pediatr Infect Dis J 2001;20:40--8.
- Gortmaker SL, Hughes M, Cervia J, et al. Effect of combination therapy including protease inhibitors on mortality among children and adolescents infected with HIV-1. N Engl J Med 2001;345:1522--8.
- Gona P, Van Dyke RB, Williams PL, et al. Incidence of opportunistic and other infections in HIV-infected children in the HAART era. JAMA 2006;296:292--300.
- Nesheim SR, Kapogiannis BG, Soe MM, et al. Trends in opportunistic infections in the pre- and post-highly active antiretroviral therapy eras among HIV-infected children in the Perinatal AIDS Collaborative Transmission Study, 1986-2004. Pediatrics 2007;120:100--9.
- Ylitalo N, Brogly S, Hughes MD, et al. Risk factors for opportunistic illnesses in children with human immunodeficiency virus in the era of highly active antiretroviral therapy. Arch Pediatr Adolesc Med 2006;160:778--87.
- CDC. USPHS/IDSA guidelines for the prevention of opportunistic infections in persons infected with human immunodeficiency virus: a summary. MMWR 1995;44(No. RR-8).
- CDC. USPHS/IDSA guidelines for the prevention of opportunistic infections in persons infected with human immunodeficiency virus. USPHS/IDSA Prevention of Opportunistic Infections Working Group. MMWR 1997;46(No. RR-12).
- CDC. 1999 USPHS/IDSA guidelines for the prevention of opportunistic infections in persons infected with human immunodeficiency virus: U.S. Public Health Service (USPHS) and Infectious Diseases Society of America (IDSA). MMWR 1999;48(No. RR-10).
- Mofenson LM, Oleske J, Serchuck L, et al. Treating opportunistic infections among HIV-exposed and infected children: recommendations from CDC, the National Institutes of Health, and the Infectious Diseases Society of America. The most recent information is available at http://aidsinfo.nih.gov. Clin Infect Dis 2005;40(Supp 1):S1--84.
- Yeung LT, King SM, Roberts EA. Mother-to-infant transmission of hepatitis C virus. Hepatology 2001;34:223--9.
- Kovacs A, Schluchter M, Easley K, et al. Cytomegalovirus infection and HIV-1 disease progression in infants born to HIV-1-infected women. Pediatric Pulmonary and Cardiovascular Complications of Vertically Transmitted HIV Infection Study Group. N Engl J Med 1999;341:77--84.
- Gutman LT, Moye J, Zimmer B, et al. Tuberculosis in human immunodeficiency virus-exposed or -infected United States children. Pediatr Infect Dis J 1994;13:963--8.
- Lindegren ML, Steinberg S, Byers R. Epidemiology of HIV/AIDS in children. Pediatr Clin North Am 2000;47:1--20.
- Working Group on Antiretroviral Therapy and Medical Management of HIV-Infected Children. Guidelines for the use of antiretroviral agents in pediatric HIV infection. February 23, 2009. Available at http://aidsinfo.nih.gov/ContentFiles/PediatricGuidelines.pdf. The most recent information is vailable at: http://aidsinfo.nih.gov.
- CDC. Guidelines for preventing opportunistic infections among HIV-infected persons---2002. Recommendations of the U.S. Public Health Service and the Infectious Diseases Society of America. MMWR 2002;51(No. RR-8).
- CDC. Guidelines for the prevention and treatment of opportunistic infections in HIV-infected adults and adolescents. Recommendations from the CDC, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. MMWR 2009;58 (No. RR-4).
- Kish MA, Infectious Diseases Society of America. Guide to development of practice guidelines. Clin Infect Dis 2001;32:851--4.
- Bohjanen PR, Boulware DR. Immune reconstitution inflammatory syndrome. In: Volberding P, Sande MA, Lange J, Greene W. eds. Global HIV/AIDS Medicine. Philadelphia, PA: Elsevier; 2007:193--205.
- Murdoch DM, Venter WD, Van Rie A, et al. Immune reconstitution inflammatory syndrome (IRIS): review of common infectious manifestations and treatment options. AIDS Res Ther 2007;4
(May 8):9. - Tangsinmankong N, Kamchaisatian W, Lujan-Zilbermann J, et al. Varicella zoster as a manifestation of immune restoration disease in HIV-infected children. J Allergy Clin Immunol 2004;113:742--6.
- Nuttall JJ, Wilmshurst JM, Ndondo AP, et al. Progressive multifocal leukoencephalopathy after initiation of highly active antiretroviral therapy in a child with advanced human immunodeficiency virus infection: a case of immune reconstitution inflammatory syndrome. Pediatr Infect Dis J 2004;23:683--5.
- Puthanakit T, Oberdorfer P, Punjaisee S, et al. Immune reconstitution syndrome due to bacillus Calmette-Guérin after initiation of antiretroviral therapy in children with HIV infection. Clin Infect Dis 2005;41:1049--52.
- Puthanakit T, Oberdorfer P, Ukarapol N, et al. Immune reconstitution syndrome from nontuberculous mycobacterial infection after initiation of antiretroviral therapy in children with HIV infection. Pediatr Infect Dis J 2006;25:645--8.
- Puthanakit T, Oberdorfer P, Akarathum N, et al. Immune reconstitution syndrome after highly active antiretroviral therapy in human immunodeficiency virus-infected thai children. Pediatr Infect Dis J 2006;25:53--8.
- Siberry GK, Tessema S. Immune reconstitution syndrome precipitated by bacille Calmette Guerin after initiation of antiretroviral therapy. Pediatr Infect Dis J 2006;25:648--9.
- Kroidl A, Huck K, Weinspach S, et al. Immune reconstitution inflammatory syndrome (IRIS) due to bacille Calmette Guerin (BCG) in an HIV-positive child. Scan J Infect Dis 2006;38:716--8.
- van Toorn R, Rabie H, Warwick JM. Opsoclonus-myoclonus in an HIV-infected child on antiretroviral therapy---possible immune reconstitution inflammatory syndrome. Eur J Paediatr Neurol 2005;9:423--6.
- Alexander A, Rode H. Adverse reactions to the Bacillus Calmette-Guerin vaccine in HIV-positive infants. J Pediatr Surg 2007;42:549--52.
- Robertson J, Meier M, Wall J, et al. Immune reconstitution syndrome in HIV: validating a case definition and identifying clinical predictors in persons initiating antiretroviral therapy. Clin Infect Dis 2006;42:1639--46.
- CDC. General recommendations on immunization: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR 2006;55(No. RR-15).
- CDC. Measles, mumps, and rubella---vaccine use and strategies for elimination of measles, rubella, and congenital rubella syndrome and control of mumps: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR 1998;47(No. RR-8).
- CDC. Prevention of varicella: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR 2007;56(No. RR-4).
- CDC. Prevention of rotavirus gastroenteritis among infants and children. Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR 2009;58(No. RR-2).
- CDC. Haemophilus b conjugate vaccines for prevention of Haemophilus influenzae type b disease among infants and children two months of age and older. Recommendations of the immunization practices advisory committee (ACIP). MMWR 1991;40(No. RR-1).
- CDC. Prevention and control of influenza. Recommendations of the Advisory Committee on Immunization Practices (ACIP), 2007. MMWR 2008;57(No. RR-7).
- CDC. Preventing pneumococcal disease among infants and young children. Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR 2000;49(No. RR-9).
- CDC. Poliomyelitis prevention in the United States. Updated recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR 2000;49(No. RR-5).
- CDC. A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP). Part 1: immunization of infants, children, and adolescents. MMWR 2005;54(No. RR-16).
- CDC. Prevention of hepatitis A through active or passive immunization: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR 2006;55(No. RR-7).
- CDC. Prevention and control of meningococcal disease. Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR 2005;54(No. RR-7).
- CDC. Notice to readers: Recommendation from the Advisory Committee on Immunization Practices (ACIP) for use of quadrivalent meningococcal conjugate vaccine (MCV4) in children aged 2--10 years at increased risk for invasive meningococcal disease. MMWR 2007:56:1265--6.
- CDC. Pertussis vaccination: use of acellular pertussis vaccines among infants and young children. Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR 1997;46(No. RR-7).
- CDC. Preventing tetanus, diphtheria, and pertussis among adolescents: use of tetanus toxoid, reduced diphtheria toxoid and acellular pertussis vaccines recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR 2006;55(No. RR-3).
- CDC. Revised recommendations of the Advisory Committee on Immunization Practices to vaccinate all persons aged 11--18 years with meningococcal conjugate vaccine. MMWR 2007;56:794--5.
- Mofenson LM, Korelitz J, Pelton S, et al. Sinusitis in children infected with human immunodeficiency virus: clinical characteristics, risk factors, and prophylaxis. National Institute of Child Health and Human Development Intravenous Immunoglobulin Clinical Trial Study Group. Clin Infect Dis 1995;21:1175--81.
- Nachman S, Gona P, Dankner W, et al. The rate of serious bacterial infections among HIV-infected children with immune reconstitution who have discontinued opportunistic infection prophylaxis. Pediatrics 2005;115:e488--94.
- Murphy TF HF, Clyde WA, Jr, Collier AM, Denny FW. Pneumonia: an eleven-year study in a pediatric practice. Am J Epidemiol 1981;113:
12--21. - Jokinen C, Heiskanen L, Juvonen H, et al. Incidence of community-acquired pneumonia in the population of four municipalities in eastern Finland. Am J Epidemiol 1993;137:977--88.
- Zangwill KM, Vadheim CM, Vannier AM, et al. Epidemiology of invasive pneumococcal disease in southern California: implications for the design and conduct of a pneumococcal conjugate vaccine efficacy trial. J Infect Dis 1996;174:752--9.
- CDC. Active bacterial core surveillance of the Emerging Infections Program Network, Streptococcus pneumoniae, 2003. Available at: www.cdc.gov/ncidod/dbmd/abcs. Accessed February 11, 2005.
- Mofenson LM, Yogev R, Korelitz J, et al. Characteristics of acute pneumonia in human immunodeficiency virus-infected children and association with long term mortality risk. National Institute of Child Health and Human Development Intravenous Immunoglobulin Clinical Trial Study Group. Pediatr Infect Dis J 1998;17:872--80.
- Madhi SA, Petersen K, Madhi A, et al. Increased disease burden and antibiotic resistance of bacteria causing severe community-acquired lower respiratory tract infections in human immunodeficiency virus type 1-infected children. Clin Infect Dis 2000;31:170--6.
- Sharland M, Gibb DM, Holland F. Respiratory morbidity from lymphocytic interstitial pneumonitis (LIP) in vertically acquired HIV infection. Arch Dis Child 1997;76:334--6.
- Intravenous immune globulin for the prevention of bacterial infections in children with symptomatic human immunodeficiency virus infection. The National Institute of Child Health and Human Developments Intravenous Immunoglobulin Study Group. N Engl J Med 1991;325:73--80.
- Spector SA, Gelber RD, McGrath N, et al. A controlled trial of intravenous immune globulin for the prevention of serious bacterial infections in children receiving zidovudine for advanced human immunodeficiency virus infection. Pediatric AIDS Clinical Trials Group. N Engl J Med 1994;331:1181--7.
- Madhi SA, Petersen K, Madhi A, et al. Impact of human immunodeficiency virus type 1 on the disease spectrum of Streptococcus pneumoniae in South African children. Pediatr Infect Dis J 2000;19:1141--7.
- Lichenstein R, King JC Jr, Farley JJ, et al. Bacteremia in febrile human immunodeficiency virus-infected children presenting to ambulatory care settings. Pediatr Infect Dis J 1998;17:381--5.
- Farley JJ, King JC Jr, Nair P, et al. Invasive pneumococcal disease among infected and uninfected children of mothers with human immunodeficiency virus infection. J Pediatr 1994;124:853--8.
- Andiman WA, Simpson J, Holtkamp C, et al. Invasive pneumococcal infections in children infected with HIV are not associated with splenic dysfunction. AIDS Patient Care STDS 1996;10:336--41.
- Mao C, Harper M, McIntosh K, et al. Invasive pneumococcal infections in human immunodeficiency virus-infected children. J Infect Dis 1996;173:870--6.
- Klugman KP, Madhi SA, Feldman C. HIV and pneumococcal disease. Curr Opin Infect Dis 2007;20:11--5.
- Crewe-Brown HH, Karstaedt AS, Saunders GL, et al. Streptococcus pneumoniae blood culture isolates from patients with and without human immunodeficiency virus infection: alterations in penicillin susceptibilities and in serogroups or serotypes. Clin Infect Dis 1997;25:1165--72.
- Klugman KP, Madhi SA, Huebner RE, et al. A trial of a 9-valent pneumococcal conjugate vaccine in children with and those without HIV infection. N Engl J Med 2003;349:1341--8.
- Frankel RE, Virata M, Hardalo C, et al. Invasive pneumococcal disease: clinical features, serotypes, and antimicrobial resistance patterns in cases involving patients with and without human immunodeficiency virus infection. Clin Infect Dis 1996;23:577--84.
- Gómez-Barreto D, Calderón-Jaimes E, Rodríguez RS, et al. Clinical outcome of invasive infections in children caused by highly penicillin-resistant Streptococcus pneumoniae compared with infections caused by penicillin-susceptible strains. Arch Med Res 2000;31:592--8.
- Rowland KE, Turnidge JD. The impact of penicillin resistance on the outcome of invasive Streptococcus pneumoniae infection in children. Aust N Z J Med 2000;30:441--9.
- Madhi SA, Petersen K, Khoosal M, et al. Reduced effectiveness of Haemophilus influenzae type b conjugate vaccine in children with a high prevalence of human immunodeficiency virus type 1 infection. Pediatr Infect Dis J 2002;21:315--21.
- Nelson CG, Iler MA, Woods CW, et al. Meningococcemia in a patient coinfected with hepatitis C virus and HIV. Emerg Infect Dis 2000;6:646--8.
- Nitta AT, Douglas JM, Arakere G, et al. Disseminated meningococcal infection in HIV-seropositive patients. AIDS 1993;7:87--90.
- Pearson IC, Baker R, Sullivan AK, et al. Meningococcal infection in patients with the human immunodeficiency virus and acquired immunodeficiency syndrome. Int J STD AIDS 2001;12:410--1.
- Kipp W, Kamugisha J, Rehle T. Meningococcal meningitis and HIV infection: results from a case-control study in western Uganda. AIDS 1992;6:1557--8.
- Stephens DS, Hajjeh RA, Baughman WS, et al. Sporadic meningococcal disease in adults: results of a 5-year population-based study. Ann Intern Med 1995;123:937--40.
- Rongkavilit C, Rodriguez ZM, Gómez-Marín O, et al. Gram-negative bacillary bacteremia in human immunodeficiency virus type 1-infected children. Pediatr Infect Dis J 2000;19:122--8.
- Berkley JA, Lowe BS, Mwangi I, et al. Bacteremia among children admitted to a rural hospital in Kenya. N Engl J Med 2005;352:39--47.
- Roilides E, Marshall D, Venzon D, et al. Bacterial infections in human immunodeficiency virus type 1-infected children: the impact of central venous catheters and antiretroviral agents. Pediatr Infect Dis J 1991;10:813--9.
- Kuhn L, Kasonde P, Sinkala M, et al. Does severity of HIV disease in HIV-infected mothers affect mortality and morbidity among their uninfected infants? Clin Infect Dis 2005;41:1654--61.
- Brahmbhatt H, Kigozi G, Wabwire-Mangen F, et al. Mortality in HIV-infected and uninfected children of HIV-infected and uninfected mothers in rural Uganda. J Acquir Immune Defic Syndr 2006;41:504--8.
- Mussi-Pinhata M, Freimanis L, Yamamoto AY, et al. Infectious disease morbidity among young HIV-1--exposed but uninfected infants in Latin American and Caribbean countries: the National Institute of Child Health and Human Development International Site Development Initiative Perinatal Study. Pediatrics 2007;119:e694--704.
- Kattan M, Platzker A, Mellins RB, et al. Respiratory diseases in the first year of life in children born to HIV-1-infected women. Pediatr Pulmonol 2001;31:267--76.
- Paul ME, Chantry CJ, Read JS, et al. Morbidity and mortality during the first two years of life among uninfected children born to human immunodeficiency virus type 1-infected women: the women and infants transmission study. Pediatr Infect Dis J 2005;24:46--56.
- Abrams EJ. Opportunistic infections and other clinical manifestations of HIV disease in children. Pediatr Clin North AM 2000;47:79--108.
- Gesner M, Desiderio D, Kim M, et al. Streptococcus pneumoniae in human immunodeficiency virus type 1-infected children. Pediatr Infect Dis J 1994;13:697--703.
- Molyneux EM, Tembo M, Kayira K, et al. The effect of HIV infection on paediatric bacterial meningitis in Blantyre, Malawi. Arch Dis Child 2003;88:1112--8.
- Madhi SA, Madhi A, Petersen K, et al. Impact of human immunodeficiency virus type 1 infection on the epidemiology and outcome of bacterial meningitis in South African children. Int J Infect Dis 2001;5:119--25.
- McIntosh K. Community-acquired pneumonia in children. N Engl J Med 2002;346:429--37.
- Zar HJ, Tannenbaum E, Hanslo D, et al. Sputum induction as a diagnostic tool for community-acquired pneumonia in infants and young children from a high HIV prevalence area. Pediatr Pulmonol 2003;36:58--62.
- Selwyn PA, Pumerantz AS, Durante A, et al. Clinical predictors of Pneumocystis carinii pneumonia, bacterial pneumonia and tuberculosis in HIV-infected patients. AIDS 1998;12:885--93.
- Sheikh S, Madiraju K, Steiner P, et al. Bronchiectasis in pediatric AIDS. Chest 1997;112:1202--7.
- Midulla F, Strappini P, Sandstrom T, et al. Cellular and noncellular components of bronchoalveolar lavage fluid in HIV-1-infected children with radiological evidence of interstitial lung damage. Pediatr Pulmonol 2001;31:205--13.
- American Academy of Pediatrics. Red book: 2006 report of the Committee on Infectious Diseases. 27th ed. Pickering LK, Baker CJ, Long SS, McMillan JA, eds. Elk Grove Village, IL;2006.
- CDC. Prevention of pneumococcal disease: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR 1997;46(No. RR-8). Available at http://www.cdc.gov/mmwr/pdf/rr/rr4608.pdf. Accessed June 13, 2009.
- Abzug MJ, Pelton SI, Song LY, et al. Immunogenicity, safety, and predictors of response after a pneumococcal conjugate and pneumococcal polysaccharide vaccine series in human immunodeficiency virus-infected children receiving highly active antiretroviral therapy. Pediatr Infect Dis J 2006;25:920--9.
- Madhi SA, Ramasamy N, Bessellar TG, et al. Lower respiratory tract infections associated with influenza A and B viruses in an area with a high prevalence of pediatric human immunodeficiency type 1 infection. Pediatr Infect Dis J 2002;21:291--7.
- Mulenga V, Ford D, Walker AS, et al. Effect of cotrimoxazole on causes of death, hospital admissions and antibiotic use in HIV-infected children. AIDS 2007;21:77--84.
- Hughes WT, Dankner WM, Yogev R, et al. Comparison of atovaquone and azithromycin with trimethoprim--sulfamethoxazole for the prevention of serious bacterial infections in children with HIV infection. Clin Infect Dis 2005;40:136--45.
- Bobat R, Coovadia H, Stephen C, et al. Safety and efficacy of zinc supplementation for children with HIV-1 infection in South Africa: a randomised double-blind placebo-controlled trial. Lancet 2005;366:1862--7.
- Irlam JH, Visser ME, Rollins N, et al. Micronutrient supplementation in children and adults with HIV infection. Cochrane Database Syst Rev 2005;Oct 19(4):CD003650.
- CDC. 1994 Revised classification system for human immunodeficiency virus infection in children less than 13 years of age. Official authorized addenda: human immunodeficiency virus infection codes and official guidelines for coding and reporting ICD-9-CM. MMWR 1994;43:1--19.
- Sáez-Llorens X, McCracken GH, Jr. Bacterial meningitis in children. Lancet 2003;361:2139--48.
- Martínez-Aguilar G, Hammerman WA, Mason EO, Jr, et al. Clindamycin treatment of invasive infections caused by community-acquired, methicillin-resistant and methicillin-susceptible Staphylococcus aureus in children. Pediatr Infect Dis J 2003;22:593--8.
- Kaplan SL, Hulten KG, Gonzalez BE, et al. Three-year surveillance of community-acquired Staphylococcus aureus infections in children. Clin Infect Dis 2005;40:1785--91.
- Moallem HJ, Garratty G, Wakeham M, et al. Ceftriaxone-related fatal hemolysis in an adolescent with perinatally acquired human immunodeficiency virus infection. J Pediatr 1998;133:279--81.
- Chintu C, Bhat GJ, Walker AS, et al. Co-trimoxazole as prophylaxis against opportunistic infections in HIV-infected Zambian children (CHAP): a double-blind randomised placebo-controlled trial. Lancet 2004;364:1865--71.
- Rolain JM, Brouqui P, Koehler JE, et al. Recommendations for treatment of human infections caused by Bartonella species. Antimicrob Agents Chemother 2004;48:1921--33.
- Wormser GP. Discovery of new infectious diseases---Bartonella species. N Engl J Med 2007;356:2346--7.
- Slater LN, Welch DF. Bartonella, including cat-scratch disease. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and practice of infectious diseases. Philadelphia, PA: Churchill Livingstone; 2005.
- Koehler JE, Quinn FD, Berger TG, et al. Isolation of Rochalimaea species from cutaneous and osseous lesions of bacillary angiomatosis. N Engl J Med 1992;327:1625--31.
- Spach DH, Koehler JE. Bartonella-associated infections. Infect Dis Clin North Am 1998;12:137--55.
- American Academy of Pediatrics. Cat-scratch disease. In: Pickering LK, Baker CJ, Long SS, McMillan JA, eds. Red book: 2006 report of the Committee on Infectious Diseases. 27th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2006.
- Reynolds MG, Holman RC, Curns AT, et al. Epidemiology of cat-scratch disease hospitalizations among children in the United States. Pediatr Infect Dis J 2005;24:700--4.
- Chomel BB, Kasten RW, Floyd-Hawkins K, et al. Experimental transmission of Bartonella henselae by the cat flea. J Clin Microbiol 1996;34:1952--6.
- Zangwill KM, Hamilton DH, Perkins BA, et al. Cat scratch disease in Connecticut. Epidemiology, risk factors, and evaluation of a new diagnostic test. N Engl J Med 1993;329:8--13.
- Carithers HA. Cat-scratch disease. An overview based on a study of 1,200 patients. Am J Dis Child 1985;139:1124--33.
- Jackson LA, Perkins BA, Wenger JD. Cat scratch disease in the United States: an analysis of three national databases. Am J Public Health 1993;83:1707--11.
- Massei F, Gori L, Macchia P, et al. The expanded spectrum of bartonellosis in children. Infect Dis Clin North Am 2005;19:691--711.
- Resto-Ruiz S, Burgess A, Anderson BE. The role of the host immune response in pathogenesis of Bartonella henselae. DNA Cell Biol 2003;22:431--40.
- Myers SA, Prose NS, Garcia JA, et al. Bacillary angiomatosis in a child undergoing chemotherapy. J Pediatr 1992;121:574--8.
- Tappero JW, Koehler JE, Berger TG, et al. Bacillary angiomatosis and bacillary splenitis in immunocompetent adults. Ann Intern Med 1993;118:363--5.
- Koehler JE, Glaser CA, Tappero JW. Rochalimaea henselae infection. A new zoonosis with the domestic cat as reservoir. JAMA 1994;271:531--5.
- Koehler JE. Bartonellosis. In: Dolin R, Masur H, Saag MS, eds. AIDS therapy. 2nd ed. New York, NY: Churchill Livingstone; 2003: 491--7.
- Koehler JE, Sanchez MA, Tye S, et al. Prevalence of Bartonella infection among human immunodeficiency virus-infected patients with fever. Clin Infect Dis 2003;27:559--66.
- Regnery RL, Childs JE, Koehler JE. Infections associated with Bartonella species in persons infected with human immunodeficiency virus. Clin Infect Dis 1995;21(Suppl 1):S94--8.
- Gerber MA. Bartonella species (cat-scratch disease, bacillary angiomatosis, bacillary peliosis). In: Long SS, Pickering LK, Prober CG, eds. Principles and practice of pediatric infectious diseases. 2nd ed. Orlando, FL: Churchill Livingstone; 2003.
- Bass JW, Vincent J, Person DA. The expanding spectrum of Bartonella infections: II. Cat-scratch disease. Pediatr Infect Dis J 1997;16:163--79.
- Sander A, Penno S. Semiquantitative species-specific detection of Bartonella henselae and Bartonella quintana by PCR-enzyme immunoassay. J Clin Microbiol 1999;37:3097--101.
- Matar GM, Koehler JE, Malcolm G, et al. Identification of Bartonella species directly in clinical specimens by PCR-restriction fragment length polymorphism analysis of a 16S rRNA gene fragment. J Clin Microbiol 1999;37:4045--7.
- Regnery R, Tappero J. Unraveling mysteries associated with cat-scratch disease, bacillary angiomatosis, and related syndromes. Emerg Infect Dis 1995;1:16--21.
- Bass JW, Freitas BC, Freitas AD, et al. Prospective randomized double blind placebo-controlled evaluation of azithromycin for treatment of cat-scratch disease. Pediatr Infect Dis J 1998;17:447--52.
- Guerra LG, Neira CJ, Boman D, et al. Rapid response of AIDS-related bacillary angiomatosis to azithromycin. Clin Infect Dis 1993;17:264--6.
- Masur H. Management of opportunistic infections associated with human immunodeficiency virus infection. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and practice of infectious diseases. 6th ed. Philadelphia, PA: Churchill Livingstone; 2005: 1679--1706.
- Gouriet F, Lepidi H, Habib G, et al. From cat scratch disease to endocarditis, the possible natural history of Bartonella henselae infection. BMC Infect Dis 2007;7(Apr 18):30.
- Conrad DA. Treatment of cat-scratch disease. Curr Opin Pediatr 2001;13:56--9.
- Sheffield JS, Sanchez PJ, Morris G, et al. Congenital syphilis after maternal treatment for syphilis during pregnancy. Am J Obstet Gynecol 2002;186:569--73.
- Alexander JM, Sheffield JS, Sanchez PJ, et al. Efficacy of treatment for syphilis in pregnancy. Obstet Gynecol 1999;93:5--8.
- CDC. Sexually transmitted disease surveillance, 2005. Atlanta, GA: U.S. Department of Health and Human Services. November 2006. Available at http://www.cdc.gov/std/stats05/05pdf/2005-exordium.pdf.
- Sison CG, Ostrea EM, Reyes MP, et al. The resurgence of congenital syphilis: a cocaine-related problem. J Pediatr 1997;130:289--92.
- Schulte JM, Burkham S, Hamaker D, et al. Syphilis among HIV-infected mothers and their infants in Texas from 1988 to 1994. Sex Transm Dis 2001;28:315--20.
- Lee MJ, Hallmark RJ, Frenkel LM, et al. Maternal syphilis and vertical perinatal transmission of human immunodeficiency virus type-1 infection. Int J Gynaecol Obstet 1998;63:247--52.
- Mwapasa V, Rogerson SJ, Kwiek JJ, et al. Maternal syphilis infection is associated with increased risk of mother-to-child transmission of HIV in Malawi. AIDS 2006;20:1869--77.
- Vermund SH, Wilson CM, Rogers AS, et al. Sexually transmitted infections among HIV infected and HIV uninfected high-risk youth in the REACH study. Reaching for Excellence in Adolescent Care and Health. J Adolesc Health 2001;29(3 Suppl):49--56.
- Blocker ME, Levine WC, St Louis ME. HIV prevalence in patients with syphilis, United States. Sex Transm Dis 2000;27:53--9.
- Singh R, McCloskey J. Syphilis in pregnancy. Venereology 2001:14:121--31.
- Michelow IC, Wendel GD Jr, Norgard MV, et al. Central nervous system infection in congenital syphilis. N Engl J Med 2002;346:1792--8.
- Glaser JH. Centers for Disease Control and Prevention guidelines for congenital syphilis. J Pediatr 1996;129:488--90.
- CDC. Symptomatic early neurosyphilis among HIV-positive men who have sex with men---four cities, United States, January 2002--June 2004. MMWR 2007;56:625--8.
- Pope V. Use of treponemal tests to screen for syphilis. Infect Med 2004;21:399--404.
- Juardo R, Campbell J, Martin PD. Prozone phenomenon in secondary syphilis. Has its time arrived? Arch Intern Med 1993;153:2496--8.
- CDC. Congenital syphilis---United States, 2002. MMWR 2004;53:716--9.
- Beltrami J, Berman S. Congenital syphilis: a persisting sentinel public health event. Sex Transm Dis 2006;33:675--6.
- Kamb ML, Fishbein M, Douglas JM, Jr, et al. Efficacy of risk-reduction counseling to prevent human immunodeficiency virus and sexually transmitted diseases: a randomized controlled trial. Project RESPECT Study Group. JAMA 1998;280:1161--7.
- Fisher JD, Cornman DH, Osborn CY, et al. Clinician-initiated HIV risk reduction intervention for HIV-positive persons: Formative Research, Acceptability, and Fidelity of the Options Project. J Acquir Immune Defic Syndr 2004;37(Suppl 2):S78--87.
- Richardson JL, Milam J, Stoyanoff S, et al. Using patient risk indicators to plan prevention strategies in the clinical care setting. J Acquir Immune Defic Syndr 2004;37(Suppl 2):S88--94.
- CDC, Health Resources and Services Administration, National Institutes of Health, et al. Recommendations for incorporating human immunodeficiency virus (HIV) prevention into the medical care of persons living with HIV. Clin Infect Dis 2004;38:104--21.
- CDC. Sexually transmitted diseases treatment guidelines, 2006. MMWR 2006;55(No. RR-11).
- Yinnon A, Coury-Doniger P, Polito R, et al. Serologic response to treatment of syphilis in patients with HIV infection. Arch Intern Med 1996;156:321--5.
- Moloney G, Branley M, Kotsiou G, et al. Syphilis presenting as scleritis in an HIV-positive man undergoing immune reconstitution. Clin Experiment Ophthalmol 2004;32:526--8.
- CDC. Trends in tuberculosis incidence---United States, 2006. MMWR 2007;56:245--50.
- Nelson LJ, Schneider E, Wells CD, et al. Epidemiology of childhood tuberculosis in the United States, 1993--2001: the need for continued vigilance. Pediatrics 2004;114:333--41.
- Coovadia HM, Jeena P, Wilkinson D. Childhood human immunodeficiency virus and tuberculosis co-infections: reconciling conflicting data. Int J Tuberc Lung Dis 1998;2:844--51.
- Thomas P, Bornschlegel K, Singh TP, et al. Tuberculosis in human immunodeficiency virus-infected and human immunodeficiency virus-exposed children in New York City. The New York City Pediatric Spectrum of HIV Disease Consortium. Pediatr Infect Dis J 2000;19:700--6.
- Shah RS, Tullu MS, Kamat JR. Clinical profile of pediatric HIV infection from India. Arch Med Res 2005;36:24--31.
- Marais BJ, Gie RP, Schaaf HS, et al. The spectrum of disease in children treated for tuberculosis in a highly endemic area. Int J Tuberc Lung Dis 2006;10:732--8.
- Adhikari M, Pillay T, Pillay DG. Tuberculosis in the newborn: an emerging disease. Pediatr Infect Dis J 1997;16:1108--12.
- Pillay T, Strum AW, Khan M, et al. Vertical transmission of Mycobacterium tuberculosis in KwaZulu Natal: impact of HIV-1 co-infection. Int J Tuberc Lung Dis 2004;8:59--69.
- Bakshi SS, Alvarez D, Hilfer CL, et al. Tuberculosis in human immunodeficiency virus-infected children. A family infection. Am J Dis Child 1993;147:320--4.
- CDC. Human tuberculosis caused by Mycobacterium bovis---New York City, 2001--2004. MMWR 2005;54:605--8.
- LoBue PA, LeClair JJ, Moser KS. Contact investigation for cases of pulmonary Mycobacterium bovis. Int J Tuberc Lung Dis 2004;8:868--72.
- Hesseling AC, Rabie H, Marais BJ, et al. Bacille Calmette-Guérin vaccine-induced disease in HIV-infected and HIV-uninfected children. Clin Infect Dis 2006;42:548--58.
- Mukadi YD, Wiktor SZ, Coulibaly IM, et al. Impact of HIV infection on the development, clinical presentation, and outcome of tuberculosis among children in Abidjan, Côte d'Ivoire. AIDS 1997;11:1151--8.
- Chintu C, Bhat G, Luo C, et al. Seroprevalence of human immunodeficiency virus type 1 infection in Zambian children with tuberculosis. Pediatr Infect Dis J 1993;12:499--504.
- Hoffman ND, Kelly C, Futterman D. Tuberculosis infection in human immunodeficiency virus-positive adolescents and young adults: a New York City cohort. Pediatrics 1996;97:198--203.
- Schaaf HS, Geldenduys A, Gie RP, et al. Culture-positive tuberculosis in human immunodeficiency virus type 1-infected children. Pediatr Infect Dis J 1998;17:599--604.
- Khouri YF, Mastrucci MT, Hutto C, et al. Mycobacterium tuberculosis in children with human immunodeficiency virus type 1 infection. Pediatr Infect Dis J 1992;11:950--5.
- Chan SP, Birnbaum J, Rao M, et al. Clinical manifestation and outcome of tuberculosis in children with acquired immunodeficiency syndrome. Pediatr Infect Dis J 1996;15:443--7.
- Starke JR. New concepts in childhood tuberculosis. Curr Opin Pediatr 2007;19:306--13.
- Maunatlala C, Alexander H, Williams KL, et al. Reproducibility of an INF-γ release assay and concordance with tuberculin skin tests in people living with HIV. Int J Tuberc Lung Dis 2007; 11 (Supplement 1): S 74. Int J Tuberc Lung Dis 2007;11(Suppl 1):S74.
- Hesseling AC, Mandalakas AM, Chegou N, et al. The impact of M. tuberculosis exposure, age and HIV infection on T-cell assays to detect M. tuberculosis infection. Int J Tuberc Lung Dis 2007;11(Suppl 1):S225.
- Starke JR. Diagnosis of tuberculosis in children. Pediatr Infect Dis J 2000;19:1095--6.
- Pomputius WF, 3rd, Rost J, Dennehy PH, et al. Standardization of gastric aspirate technique improves yield in the diagnosis of tuberculosis in children. Pediatr Infect Dis J 1997;16:222--6.
- Iriso R, Mudido PM, Karamagi C, et al. The diagnosis of childhood tuberculosis in an HIV-endemic setting and the use of induced sputum. Int J Tuberc Lung Dis 2005;9:716--26.
- Zar HJ, Hanslo D, Apolles P, et al. Induced sputum versus gastric lavage for microbiological confirmation of pulmonary tuberculosis in infants and young children: a prospective study. Lancet 2005;365:130--4.
- Owens S, Abdel-Rahman IE, Balyejusa S, et al. Nasopharyngeal aspiration for diagnosis of pulmonary tuberculosis. Arch Dis Child 2007;92:693--6.
- Delacourt C, Poveda JD, Chureau C, et al. Use of polymerase chain reaction for improved diagnosis of tuberculosis in children. J Pediatr 1995;126(5 Pt 1):703--9.
- Pierre C, Olivier C, Lecossier D, et al. Diagnosis of primary tuberculosis in children by amplification and detection of mycobacterial DNA. Am Rev Respir Dis 1993;147:420--4.
- Smith K, Starke J, Eisenach K, et al. Detection of Mycobacterium tuberculosis in clinical specimens from children using a polymerase chain reaction. Pediatrics 1996;97:155--60.
- World Health Organization. Guidance for national tuberculosis programmes on the management of tuberculosis in children. Geneva. WHO/HTM/TB/2006.371. 2006.
- Schaaf HS, Gie RP, Beyers N, et al. Primary drug-resistant tuberculosis in children. Int J Tuberc Lung Dis 2000;4:1149--55.
- Schaaf HS, Gie RP, Kennedy M, et al. Evaluation of young children in contact with adult multidrug-resistant pulmonary tuberculosis: a 30-month follow-up. Pediatrics 2002;109:765--71.
- World Health Organization. Revised BCG vaccination guidelines for infants at risk for HIV infection. Wkly Epidemiol Rec 2007;82:193--6.
- Zar HJ, Cotton MF, Strauss S, et al. Effect of isoniazid prophylaxis on mortality and incidence of tuberculosis in children with HIV: randomised controlled trial. BMJ 2007;334:136.
- CDC. Acquired rifamycin resistance in persons with advanced HIV disease being treated for active tuberculosis with intermittent rifamycin-based regimens. MMWR 2002;51:214--5.
- Starke JR, Correa AG. Management of mycobacterial infection and disease in children. Pediatr Infect Dis J 1995;14:455--69.
- World Health Organization. Guidelines for the programmatic management of drug-resistant tuberculosis. Geneva, Switzerland: (WHO/HTM/TB/2006.361). 2006.
- Raviglione MC, Smith IM. XDR tuberculosis---implications for global public health. N Engl J Med 2007;356:656--9.
- CDC. Extensively drug-resistant tuberculosis---United States, 1993--2006. MMWR 2007;56:250--3.
- Palusci VJ, O'Hare D, Lawrence RM. Hepatotoxicity and transaminase measurement during isoniazid chemoprophylaxis in children. Pediatr Infect Dis J 1995;14:144--8.
- Narita M, Ashkin D, Hollender ES, et al. Paradoxical worsening of tuberculosis following antiretroviral therapy in patients with AIDS. Am J Respir Crit Care Med 1998;158:157--61.
- Wendel KA, Alwood KS, Gachuhi R, et al. Paradoxical worsening of tuberculosis in HIV-infected persons. Chest 2001;120:193--7.
- Chien JW, Johnson JL. Paradoxical reactions in HIV and pulmonary TB. Chest 1998;114:933--6.
- Zampoli M, Kilborn T, Eley B. Tuberculosis during early antiretroviral-induced immune reconstitution in HIV-infected children. Int J Tuberc Lung Dis 2007;11:417--23.
- Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med 2007;175:367--416.
- Perez Mato S, van Dyke RB. Pulmonary infections in children with HIV infection. Semin Respir Infect 2002;17:33--46.
- Reed C, von Reyn CF, Chamblee S, et al. Environmental risk factors for infection with Mycobacterium avium complex. Am J Epidemiol 2006;164:32--40.
- Peacock KH, Lewis L, Lavoie S. Erosive mediastinal lymphadenitis associated with Mycobacterium avium infection in a pediatric acquired immunodeficiency syndrome patient. Pediatr Infect Dis J 2000;19:576--8.
- Hartmann P, Plum G. Immunological defense mechanisms in tuberculosis and MAC-infection. Diagn Microbiol Infect Dis 1999;34:147--52.
- Keller C, Kirkpatrick S, Lee K, et al. Disseminated Mycobacterium avium complex presenting as hematochezia in an infant with rapidly progressive acquired immunodeficiency syndrome. Pediatr Infect Dis J 1996;15:713--5.
- Wong DA, Yip PC, Cheung DT, et al. Simple and rational approach to the identification of Mycobacterium tuberculosis, Mycobacterium avium complex species, and other commonly isolated mycobacteria. J Clin Microbiol 2001;39:3768--71.
- Lewis LL, Butler KM, Husson RN, et al. Defining the population of human immunodeficiency virus-infected children at risk for Mycobacterium avium-intracellulare infection. J Pediatr 1992;121:677--83.
- Rutstein R, Cobb P, McGowan K, et al. Mycobacterium avium intracellulare complex infection in HIV-infected children AIDS 1993;7:507--12.
- Smith JA, Mueller BU, Nussenblatt RB, Whitcup SM. Corneal endothelial deposits in children positive for human immunodeficiency virus receiving rifabutin prophylaxis for Mycobacterium avium complex bacteremia. Am J Ophthalmol 1999;127:164--9.
- Race EM, Adelson-Mitty J, Kriegel GR, et al. Focal mycobacterial lymphadenitis following initiation of protease-inhibitor therapy in patients with advanced HIV-1 disease. Lancet 1998;351:252--5.
- Phillips P, Chan K, Hogg R, et al. Azithromycin prophylaxis for Mycobacterium avium complex during the era of highly active antiretroviral therapy: evaluation of a provincial program. Clin Infect Dis 2002;34:371--8.
- Steenhoff AP, Wood SM, Shah SS, et al. Cutaneous Mycobacterium avium complex infection as a manifestation of the immune reconstitution syndrome in a human immunodeficiency virus-infected child. Pediatr Infect Dis J 2007;26:755--7.
- Long SS. Principles and practice of pediatric infectious diseases. 2nd ed. Orlando, FL: Churchill Livingstone; 2003.
- Denning DW. Invasive aspergillosis. Clin Infect Dis 1998;26:781--803.
- Herbrecht R, Auvrignon A, Andrès E, et al. Efficacy of amphotericin B lipid complex in the treatment of invasive fungal infections in immunosuppressed paediatric patients. Eur J Clin Microbiol Infect Dis 2001;20:77--82.
- Shetty D, Giri N, Gonzalez CE, et al. Invasive aspergillosis in human immunodeficiency virus-infected children. Pediatr Infect Dis J 1997;16:216--21.
- Drut R, Anderson V, Greco MA, et al. Opportunistic infections in pediatric HIV infection: a study of 74 autopsy cases from Latin America. The Latin American AIDS Pathology Study Group. Pediatr Pathol Lab Med 1997;17:569--76.
- Reik RA, Rodriguez MM, Hensley GT. Infections in children with human immunodeficiency virus/acquired immunodeficiency syndrome: an autopsy study of 30 cases in south Florida, 1990--1993. Pediatr Pathol Lab Med 1995;15:269--81.
- Denning DW, Follansbee SE, Scolaro M, et al. Pulmonary aspergillosis in the acquired immunodeficiency syndrome. N Engl J Med 1991;324:654--62.
- Groll AH, Shah PM, Mentzel C, et al. Trends in the postmortem epidemiology of invasive fungal infections at a university hospital. J Infect 1996;33:23--32.
- Lortholary O, Meyohas MC, Dupont B, et al. Invasive aspergillosis in patients with acquired immunodeficiency syndrome: report of 33 cases. French Cooperative Study Group on Aspergillosis in AIDS. Am J Med 1993;95:177--87.
- Walsh TJ, Gonzalez C, Lyman CA, et al. Invasive fungal infections in children: recent advances in diagnosis and treatment. Adv Pediatr Infect Dis 1996:11:187--290.
- Roilides E, Holmes A, Blake C, et al. Defective antifungal activity of monocyte-derived macrophages from human immunodeficiency virus-infected children against Aspergillus fumigatus. J Infect Dis 1993;168:1562--5.
- Muller FM, Trusen A, Weig M. Clinical manifestations and diagnosis of invasive aspergillosis in immunocompromised children. Eur J Pediatr 2002;161:563--74.
- Domachowske JB. Pediatric human immunodeficiency virus infection. Clin Microbiol Rev 1996;9:448--68.
- Wright M, Fikrig S, Haller JO. Aspergillosis in children with acquired immune deficiency. Pediatr Radiol 1993;23:492--4.
- Leibovitz E, Rigaud M, Chandwani S, et al. Disseminated fungal infections in children infected with human immunodeficiency virus. Pediatr Infect Dis J 1991;10:888--94.
- CDC. Treating opportunistic infections among HIV-exposed and infected children: recommendations from CDC, the National Institutes of Health, and the Infectious Diseases Society of America. The most recent information is available at http://aidsinfo.nih.gov. MMWR 2004;53(No. RR-15).
- Sulahian A, Tabouret M, Ribaud P, et al. Comparison of an enzyme immunoassay and latex agglutination test for detection of galactomannan in the diagnosis of invasive aspergillosis. Eur J Clin Microbiol Infect Dis 1996;15:139--45.
- Herbrecht R, Letscher-Bru V, Oprea C, et al. Aspergillus galactomannan detection in the diagnosis of invasive aspergillosis in cancer patients. J Clin Oncol 2002;20:1898--906.
- Greene RE, Schlamm HT, Oestmann JW, et al. Imaging findings in acute invasive pulmonary aspergillosis: clinical significance of the halo sign. Clin Infect Dis 2007;44:373--9.
- Steinbach WJ. Pediatric aspergillosis: disease and treatment differences in children. Pediatr Infect Dis J 2005;24:358--64.
- Thomas KE, Owens CM, Veys PA, et al. The radiological spectrum of invasive aspergillosis in children: a 10-year review. Pediatr Radiol 2003;33:453--60.
- Allan BT, Patton D, Ramsey NK, et al. Pulmonary fungal infections after bone marrow transplantation. Pediatr Radiol 1988;18:118--22.
- Taccone A, Occhi M, Garaventa A, et al. CT of invasive pulmonary aspergillosis in children with cancer. Pediatr Radiol 1993;23:177--80.
- Allo MD, Miller J, Townsend T, et al. Primary cutaneous aspergillosis associated with Hickman intravenous catheters. N Engl J Med 1987;317:1105--8.
- Bryce EA, Walker M, Scharf S, et al. An outbreak of cutaneous aspergillosis in a tertiary-care hospital. Infect Control Hosp Epidemiol 1996;17:170--2.
- James MJ, Lasker BA, McNeil MM, et al. Use of a repetitive DNA probe to type clinical and environmental isolates of Aspergillus flavus from a cluster of cutaneous infections in a neonatal intensive care unit. J Clin Microbiol 2000;38:3612--8.
- Ruutu P, Valtonen V, Tiitanen L, et al. An outbreak of invasive aspergillosis in a haematologic unit. Scand J Infect Dis 1987;19:347--51.
- Anaissie EJ, Stratton SL, Dignani MC, et al. Pathogenic Aspergillus species recovered from a hospital water system: a 3-year prospective study. Clin Infect Dis 2002;34:780--9.
- Opal S, Asp AA, Cannady PB, et al. Efficacy of infection control measures during a nosocomial outbreak of disseminated aspergillosis associated with hospital construction. J Infect Dis 1986;153:634--7.
- Morgenstern GR, Prentice AG, Prentice HG, et al. A randomized controlled trial of itraconazole versus fluconazole for the prevention of fungal infections in patients with haematological malignancies. U.K. Multicentre Antifungal Prophylaxis Study Group. Br J Haematol 1999;105:901--11.
- Rousey SR, Russler S, Gottlieb M, et al. Low-dose amphotericin B prophylaxis against invasive Aspergillus infections in allogeneic marrow transplantation. Am J Med 1991;91:484--92.
- Siwek GT, Pfaller MA, Polgreen PM, et al. Incidence of invasive aspergillosis among allogeneic hematopoietic stem cell transplant patients receiving voriconazole prophylaxis. Diagn Microbiol Infect Dis 2006;55:209--12.
- Herbrecht R, Denning DW, Patterson TF, et al. Voriconazole versus amphotericin B for primary therapy of invasive aspergillosis. N Engl J Med 2002;347:408--15.
- Cesaro S, Strugo L, Alaggio R, et al. Voriconazole for invasive aspergillosis in oncohematological patients: a single-center pediatric experience. Support Care Cancer 2003;11:722--7.
- Steinbach WJ. Antifungal agents in children. Pediatr Clin North Am 2005;52:895--915.
- Blyth CC, Palasanthiran P, O'Brien TA. Antifungal therapy in children with invasive fungal infections: a systematic review. Pediatrics 2007;119:772--84.
- Pannaraj PS, Walsh TJ, Baker CJ. Advances in antifungal therapy. Pediatr Infect Dis J 2005;24:921--2.
- Walsh TJ, Lutsar I, Driscoll T, et al. Voriconazole in the treatment of aspergillosis, scedosporiosis and other invasive fungal infections in children. Pediatr Infect Dis J 2002;21:240--8.
- Scott LJ, Simpson D. Voriconazole: a review of its use in the management of invasive fungal infections. Drugs 2007;67:269--98.
- Walsh TJ, Karlsson MO, Driscoll T, et al. Pharmacokinetics and safety of intravenous voriconazole in children after single- or multiple-dose administration. Antimicrob Agents Chemother 2004;48:2166--72.
- Walsh TJ, Seibel NL, Arndt C, et al. Amphotericin B lipid complex in pediatric patients with invasive fungal infections. Pediatr Infect Dis J 1999;18:702--8.
- Sambatakou H, Denning DW. Invasive pulmonary aspergillosis transformed into fatal mucous impaction by immune reconstitution in an AIDS patient. Eur J Clin Microbiol Infect Dis 2005;24:628--33.
- Walsh TJ, Adamson PC, Seibel NL, et al. Pharmacokinetics, safety, and tolerability of caspofungin in children and adolescents. Antimicrob Agents Chemother 2005;49:4536--45.
- Marr KA, Boeckh M, Carter RA, et al. Combination antifungal therapy for invasive aspergillosis. Clin Infect Dis 2004;39:797--802.
- Merlin E, Galambrun C, Ribaud P, et al. Efficacy and safety of caspofungin therapy in children with invasive fungal infections. Pediatr Infect Dis J 2006;25:1186--8.
- Cesaro S, Giacchino M, Locatelli F, et al. Safety and efficacy of a caspofungin-based combination therapy for treatment of proven or probable aspergillosis in pediatric hematological patients. BMC Infect Dis 2007;7(Apr 18):28.
- Singh N, Limaye AP, Forrest G, et al. Combination of voriconazole and caspofungin as primary therapy for invasive aspergillosis in solid organ transplant recipients: a prospective, multicenter, observational study. Transplantation 2006;81:320--6.
- Johnson MD, Perfect JR. Combination antifungal therapy: what can and should we expect? Bone Marrow Transplant 2007;40:297--306.
- Baddley JW, Pappas PG. Combination antifungal therapy for the treatment of invasive yeast and mold infections. Curr Infect Dis Rep 2007;9:448--56.
- Raad II, Hanna HA, Boktour M, et al. Novel antifungal agents as salvage therapy for invasive aspergillosis in patients with hematologic malignancies: posaconazole compared with high-dose lipid formulations of amphotericin B alone or in combination with caspofungin. Leukemia 2008;22:496--503.
- Denning DW, Marr KA, Lau WM, et al. Micafungin (FK463), alone or in combination with other systemic antifungal agents, for the treatment of acute invasive aspergillosis. J Infect 2006;53:337--49.
- Karp JE, Burch PA, Merz WG. An approach to intensive antileukemia therapy in patients with previous invasive aspergillosis. Am J Med 1988;85:203--6.
- Chiou CC, Groll AH, Gonzalez CE, et al. Esophageal candidiasis in pediatric acquired immunodeficiency syndrome: clinical manifestations and risk factors. Pediatr Infect Dis J 2000;19:729--34.
- Walsh TJ, Gonzalez C, Roilides E, et al. Fungemia in children infected with the human immunodeficiency virus: new epidemiologic patterns, emerging pathogens, and improved outcome with antifungal therapy. Clin Infect Dis 1995;20:900--6.
- Chiou CC, Groll AH, Mavrogiorgos N, et al. Esophageal candidiasis in human immunodeficiency virus-infected pediatric patients after the introduction of highly active antiretroviral therapy. Pediatr Infect Dis J 2002;21:388--92.
- Gonzalez CE, Venzon D, Lee S, et al. Risk factors for fungemia in children infected with human immunodeficiency virus: a case-control study. Clin Infect Dis 1996;23:515--21.
- Krcmery V, Augustinova A, Babelova O, et al. Fungal resistance in Cambodian children with acquired immunodeficiency syndrome. Pediatr Infect Dis J 2006;25:470.
- Muller FM, Groll AH, Walsh TJ. Current approaches to diagnosis and treatment of fungal infections in children infected with human immuno deficiency virus. Eur J Pediatr 1999;158:187--99.
- Stevens D. Diagnosis of fungal infections: current status. J Antimicrob Chemother 2002;49(Suppl 1):11--9.
- Sigmundsdottir G, Larsson L, Wiebe T, et al. Clinical experience of urine D-arabinitol/L-arabinitol ratio in the early diagnosis of invasive candidiasis in paediatric high risk populations. Scand J Infect Dis 2007;39:146--51.
- Yeo SF, Huie S, Sofair AN, et al. Measurement of serum D-arabinitol/creatinine ratios for initial diagnosis and for predicting outcome in an unselected, population-based sample of patients with Candida fungemia. J Clin Microbiol 2006;44:3894--9.
- Verduyn Lunel FM, Voss A, Kuijper EJ, et al. Detection of the Candida antigen mannan in cerebrospinal fluid specimens from patients suspected of having Candida meningitis. J Clin Microbiol 2004;42:867--70.
- Ostrosky-Zeichner L, Alexander BD, Kett DH, et al. Multicenter clinical evaluation of the (1-->3) beta-D-glucan assay as an aid to diagnosis of fungal infections in humans. Clin Infect Dis 2005;41:654--9.
- Klingspor L, Jalal S. Molecular detection and identification of Candida and Aspergillus spp. from clinical samples using real-time PCR. Clin Microbiol Infect 2006;12:745--53.
- Pienaar ED, Young T, Holmes H. Interventions for the prevention and management of oropharyngeal candidiasis associated with HIV infection in adults and children. Cochrane Database Syst Rev 2006: July 19;3:C003940.
- Rex JH, Walsh TJ, Sobel JD, et al. Practice guidelines for the treatment of candidiasis. Infectious Diseases Society of America. Clin Infect Dis 2000;30:662--78.
- Pelletier R, Peter J, Antin C, et al. Emergence of resistance of Candida albicans to clotrimazole in human immunodeficiency virus-infected children: in vitro and clinical correlations. J Clin Microbiol 2000;38:1563--8.
- Pons V, Greenspan D, Debruin M. Therapy for oropharyngeal candidiasis in HIV-infected patients: a randomized, prospective multicenter study of oral fluconazole versus clotrimazole troches. The Multicenter Study Group. J Acquir Immune Defic Syndr Hum Retrovirol 1993;6:1311--6.
- Pons V, Greenspan D, Lozada-Nur F, et al. Oropharyngeal candidiasis in patients with AIDS: randomized comparison of fluconazole versus nystatin oral suspensions. Clin Infect Dis 1997;24:1204--7.
- Goins RA, Ascher D, Waecker N, et al. Comparison of fluconazole and nystatin oral suspensions for treatment of oral candidiasis in infants. Pediatr Infect Dis J 2002;21:1165--7.
- Phillips P, de Beule K, Frechette G, et al. A double-blind comparison of itraconazole oral solution and fluconazole capsules for the treatment of oropharyngeal candidiasis in patients with AIDS Clin Infect Dis 1998;26:1368--73.
- Wilcox CM, Darouiche RO, Laine L, et al. A randomized, double-blind comparison of itraconazole oral solution and fluconazole tablets in the treatment of esophageal candidiasis. J infect Dis 1997;176:227--32.
- Mora-Duarte J, Betts R, Rotstein C, et al. Comparison of caspofungin and amphotericin B for invasive candidiasis. N Engl J Med 2002;347:2020--9.
- Walsh TJ. Echinocandins---an advance in the primary treatment of invasive candidiasis N Engl J Med 2002;347:2070--2.
- Deresinski SC, Stevens DA. Caspofungin. Clin Infect Dis 2003;36:1445--57.
- Dismukes WE. Introduction to antifungal drugs. Clin Infect Dis 2000;30:653--7.
- Brammer KW, Coates PE. Pharmacokinetics of fluconazole in pediatric patients. Eur J Clin Microbiol Infect Dis 1994;13:325--9.
- de Wet N, Llanos-Cuentas A, Suleiman J, et al. A randomized, double-blind, parallel-group, dose-response study of micafungin compared with fluconazole for the treatment of esophageal candidiasis in HIV-positive patients. Clin Infect Dis 2004;39:842--9.
- Krause DS, Simjee AE, van Rensburg C, et al. A randomized, double-blind trial of anidulafungin versus fluconazole for the treatment of esophageal candidiasis. Clin Infect Dis 2004;39:770--5.
- Villanueva A, Gotuzzo E, Arathoon EG, et al. A randomized double-blind study of caspofungin versus fluconazole for the treatment of esophageal candidiasis. Am J Med 2002;113:294--9.
- Odio CM, Araya R, Pinto LE, et al. Caspofungin therapy of neonates with invasive candidiasis. Pediatr Infect Dis J 2004;23:1093--7.
- Seibel NL, Schwartz C, Arrieta A, et al. Safety, tolerability, and pharmacokinetics of Micafungin (FK463) in febrile neutropenic pediatric patients. Antimicrob Agents Chemother 2005;49:3317--24.
- Heresi GP, Gerstmann DR, Reed MD, et al. The pharmacokinetics and safety of micafungin, a novel echinocandin, in premature infants. Pediatr Infect Dis J 2006;25:1110--5.
- Tabata K, Katashima M, Kawamura A, et al. Linear pharmacokinetics of micafungin and its active metabolites in Japanese pediatric patients with fungal infections. Biol Pharm Bull 2006;29:1706--11.
- Benjamin DK, Jr, Driscoll T, Seibel NL, et al. Safety and pharmacokinetics of intravenous anidulafungin in children with neutropenia at high risk for invasive fungal infections. Antimicrob Agents Chemother 2006;50:632--8.
- Rex JH, Pappas PG, Karchmer AW, et al. A randomized and blinded multicenter trial of high-dose fluconazole plus placebo versus fluconazole plus amphotericin B as therapy for candidemia and its consequences in nonneutropenic subjects. Clin Infect Dis 2003;36:1221--8.
- Johnson LB, Kauffman CA. Voriconazole: a new triazole antifungal agent. Clin Infect Dis 2003;36:630--7.
- Nacher M, Vantilcke V, Huber F, et al. Increased incidence of mucosal candidiasis after HAART initiation: a benign form of immune reconstitution disease? AIDS 2007;21:2534--6.
- Groll AH, Wood L, Roden M, et al. Safety, pharmacokinetics, and pharmacodynamics of cyclodextrin itraconazole in pediatric patients with oropharyngeal candidiasis. Antimicrob Agents Chemother 2002;46:2554--63.
- Phillips P, Zemcov J, Mahmood W, et al. Itraconazole cyclodextrin solution for fluconazole-refractory oropharyngeal candidiasis in AIDS: correlation of clinical response with in vitro susceptibility. AIDS 1996;10:1369--76.
- Fichtenbaum CJ, Powderly WG. Refractory mucosal candidiasis in patients with human immunodeficiency virus infection. Clin Infect Dis 1998;26:556--65.
- Skiest D, Vazquez J, Anstead G, et al. Posaconazole for the treatment of azole-refractory oropharyngeal and esophageal candidiasis in subjects with HIV infection. Clin Infect Dis 2007;44:607--14.
- Zaoutis TE, Benjamin DK, Steinbach WJ. Antifungal treatment in pediatric patients. Drug Resist Updat 2005;8:235--45.
- Krishna G, Sansone-Parsons A, Martinho M, et al. Posaconazole plasma concentrations in juvenile patients with invasive fungal infection. Antimicrob Agents Chemother 2007;51:812--8.
- Lake DE, Kunzweiler J, Beer M, et al. Fluconazole versus amphotericin B in the treatment of esophageal candidiasis in cancer patients. Chemotherapy 1996;42:308--14.
- Rex JH, Rinaldi MG, Pfaller MA. Resistance of Candida species to fluconazole. Antimicrob Agents Chemother 1995;39:1--8.
- Walsh TJ, Whitcomb P, Piscitelli S, et al. Safety, tolerance, and pharmacokinetics of amphotericin B lipid complex in children with hepatosplenic candidiasis. Antimicrob Agents Chemother 1997;41:1944--8.
- Wiley JM, Seibel NL, Walsh TJ. Efficacy and safety of amphotericin B lipid complex in 548 children and adolescents with invasive fungal infections. Pediatr Infect Dis J 2005;24:167--74.
- Walsh TJ, Finberg RW, Arndt C, et al. Liposomal amphotericin B for empirical therapy in patients with persistent fever and neutropenia. National Institute of Allergy and Infectious Diseases Mycoses Study Group. N Engl J Med 1999;340:764--71.
- Linden P, Lee L, Walsh TJ. Retrospective analysis of the dosage of amphotericin B lipid complex for the treatment of invasive fungal infections. Pharmacotherapy 1999;19:1261--8.
- Tollemar J, Klingspor L, Ringdén O. Liposomal amphotericin B (AmBisome) for fungal infections in immunocompromised adults and children. Clin Microbiol Infect 2001;7(Suppl 2):68--79.
- Goldman M, Cloud GA, Wade KD, et al. A randomized study of the use of fluconazole in continuous versus episodic therapy in patients with advanced HIV infection and a history of oropharyngeal candidiasis: AIDS Clinical Trials Group Study 323/Mycoses Study Group Study 40. Clin Infect Dis 2005;41:1473--80.
- DiCaudo DJ. Coccidioidomycosis: a review and update. J Am Acad Dermatol 2006;55:929--42.
- Crum NF, Lederman ER, Stafford CM, et al. Coccidioidomycosis: a descriptive survey of a reemerging disease. Clinical characteristics and current controversies. Medicine (Baltimore) 2004;83:149--75.
- Stagliano D, Epstein J, Hickey P. Fomite-transmitted coccidioidomycosis in an immunocompromised child. Pediatr Infect Dis J 2007;26:454--6.
- Ampel NM, Dols CL, Galgiani JN. Coccidioidomycosis during human immunodeficiency virus infection: results of a prospective study in a coccidioidal endemic area. Am J Med 1993;94:235--40.
- Ampel NM. Coccidioidomycosis in persons infected with HIV-1. Ann N Y Acad Sci 2007: 111:336--42.
- Ampel NM. Coccidioidomycosis among persons with human immunodeficiency virus infection in the era of highly active antiretroviral therapy (HAART). Semin Respir Infect 2001;16:257--62.
- Ampel NM. Coccidioidomycosis in persons infected with HIV type 1. Clin Infect Dis 2005;41:1174--8.
- Woods CW, McRill C, Plikaytis BD, et al. Coccidioidomycosis in human immunodeficiency virus-infected persons in Arizona, 1994-1997: incidence, risk factors, and prevention. J Infect Dis 2000;181:1428--34.
- Galgiani JN, Ampel NM, Catanzaro A, et al. Practice guideline for the treatment of coccidioidomycosis. Infectious Diseases Society of America. Clin Infect Dis 2000;30:658--61.
- Galgiani JN, Catanzaro A, Cloud GA, et al. Comparison of oral fluconazole and itraconazole for progressive, nonmeningeal coccidioidomycosis. A randomized, double-blind trial. Mycoses Study Group. Ann Intern Med 2000;133:676--86.
- Saitoh A, Homans J, Kovacs A. Fluconazole treatment of coccidioidal meningitis in children: two case reports and a review of the literature. Pediatr Infect Dis J 2000;19:1204--8.
- Huang YW, Chang CC, Sun HY, et al. Primary adrenal insufficiency in patients with acquired immunodeficiency syndrome: report of four cases. J Microbiol Immunol Infect 2004;37:250--3.
- Anstead GM, Corcoran G, Lewis J, et al. Refractory coccidioidomycosis treated with posaconazole. Clin Infect Dis 2005;40:1770--6.
- Proia LA, Tenorio AR. Successful use of voriconazole for treatment of Coccidioides meningitis. Antimicrob Agents Chemother 2004;48:2341.
- Prabhu RM, Bonnell M, Currier BL, et al. Successful treatment of disseminated nonmeningeal coccidioidomycosis with voriconazole. Clin Infect Dis 2004;39:e74--7.
- Hsue G, Napier JT, Prince RA, et al. Treatment of meningeal coccidioidomycosis with caspofungin. J Antimicrob Chemother 2004;54:292--4.
- Antony S. Use of the echinocandins (caspofungin) in the treatment of disseminated coccidioidomycosis in a renal transplant recipient. Clin Infect Dis 2004;39:879--80.
- Park DW, Sohn JW, Cheong HJ, et al. Combination therapy of disseminated coccidioidomycosis with caspofungin and fluconazole. BMC Infect Dis 2006: Feb 15;6:26.
- Kuberski TT, Servi RJ, Rubin PJ. Successful treatment of a critically ill patient with disseminated coccidioidomycosis, using adjunctive interferon-gamma. Clin Infect Dis 2004;38:910--2.
- Galgiani JN, Ampel NM, Blair JE, et al. Coccidioidomycosis. Clin Infect Dis 2005;41:1217--23.
- Carmichael JK. Coccidioidomycosis in HIV-infected persons. Clin Infect Dis 2006;42:1059; author reply 1059--60.
- Mathew G, Smedema M, Wheat LJ, et al. Relapse of coccidioidomycosis despite immune reconstitution after fluconazole secondary prophylaxis in a patient with AIDS. Mycoses 2003;46:42--4.
- Dewsnup DH, Galgiani JN, Graybill JR, et al. Is it ever safe to stop azole therapy for Coccidioides immitis meningitis? Ann Intern Med 1996;124:305--10.
- Leggiadro RJ, Kline MW, Hughes WT. Extrapulmonary cryptococcosis in children with acquired immunodeficiency syndrome. Pediatr Infect Dis J 1991;10:658--62.
- Gonzalez CE, Shetty D, Lewis LL, et al. Cryptococcosis in human immunodeficiency virus-infected children. Pediatr Infect Dis J 1996;15:796--800.
- Abadi J, Nachman S KA, Pirofski L. Cryptococcosis in children with AIDS. Clin Infect Dis 1999;28:309--13.
- Mirza S, Phelan M, Rimland D, et al. The changing epidemiology of cryptococcosis: an update from population-based active surveillance in 2 large metropolitan areas, 1992-2000. Clin Infect Dis 2003;36:789--94.
- Dromer F, Mathoulin-Pélissier S, Fontanet A, et al. Epidemiology of HIV-associated cryptococcosis in France (1985-2001): comparison of the pre- and post-HAART eras. AIDS 2004;18:555--62.
- Gumbo T, Kadzirange G, Mielke J, et al. Cryptococcus neoformans meningoencephalitis in African children with acquired immunodeficiency syndrome. Pediatr Infect Dis J 2002;21:54--6.
- Chuck SL, Sande MA. Infections with Cryptococcus neoformans in the acquired immunodeficiency syndrome. N Engl J Med 1989;321:794--9.
- Currie BP, Freundlich LF, Soto MA, et al. False-negative cerebrospinal fluid cryptococcal latex agglutination tests for patients with culture-positive cryptococcal meningitis. J Clin Microbiol 1993;31:2519--22.
- Pfaller MA, Messer SA, Boyken L, et al. Global trends in the antifungal susceptibility of Cryptococcus neoformans (1990 to 2004). J Clin Microbiol 2005;43:2163--7.
- Goldman DL, Khine H, Abadi J, et al. Serologic evidence for Cryptococcus neoformans infection in early childhood. Pediatrics 2001;107:E66.
- Chang LW, Phipps WT, Kennedy GE, et al. Antifungal interventions for the primary prevention of cryptococcal disease in adults with HIV. Cochrane Database Syst Rev 2005;Jul 20:CD004773.
- Shelburne SA, Darcourt J, White AC, et al. The role of immune reconstitution inflammatory syndrome in AIDS-related Cryptococcus neoformans disease in the era of highly active antiretroviral therapy. Clin Infect Dis 2005;40:1049--52.
- Shelburne SA, Visnegarwala F, Darcourt J, et al. Incidence and risk factors for immune reconstitution inflammatory syndrome during highly active antiretroviral therapy. AIDS 2005;194.
- Perfect JR, Marr KA, Walsh TJ, et al. Voriconazole treatment for less-common, emerging, or refractory fungal infections. Clin Infect Dis 2003;36:1122--31.
- Pitisuttithum P, Negroni R, Graybill JR, et al. Activity of posaconazole in the treatment of central nervous system fungal infections. J Antimicrob Chemother 2005;56:745--55.
- Bennett JE, Dismukes WE, Duma RJ, et al. A comparison of amphotericin B alone and combined with flucytosine in the treatment of cryptoccal meningitis. N Engl J Med 1979;301:126--31.
- van der Horst CM, Saag MS, Cloud GA, et al. Treatment of cryptococcal meningitis associated with the acquired immunodeficiency syndrome. National Institute of Allergy and Infectious Diseases Mycoses Study Group and AIDS Clinical Trials Group. N Engl J Med 1997;337:15--21.
- Saag MS, Graybill RJ, Larsen RA, et al. Practice guidelines for the management of cryptococcal disease. Infectious Diseases Society of America. Clin Infect Dis 2000;30:710--8.
- Brouwer AE, Rajanuwong A, Chierakul W, et al. Combination antifungal therapies for HIV-associated cryptococcal meningitis: a randomised trial. Lancet 2004;363:1764--7.
- Dromer F, Mathoulin-Pélissier S, Launay O, et al. Determinants of disease presentation and outcome during cryptococcosis: the CryptoA/D study. PLOS Med 2007;4:e21.
- Leenders AC, Reiss P, Portegies P, et al. Liposomal amphotericin B (AmBisome) compared with amphotericin B both followed by oral fluconazole in the treatment of AIDS-associated cryptococcal meningitis. AIDS 1997;11:1463--71.
- Larsen RA, Bozzette SA, Jones BE, et al. Fluconazole combined with flucytosine for treatment of cryptococcal meningitis in patients with AIDS. Clin Infect Dis 1994;19:741--5.
- Mayanja-Kizza H, Oishi K, Mitarai S, et al. Combination therapy with fluconazole and flucytosine for cryptococcal meningitis in Ugandan patients with AIDS. Clin Infect Dis 1998;26:1362--6.
- Saag MS, Powderly WG, Cloud GA, et al. Comparison of amphotericin B with fluconazole in the treatment of acute AIDS-associated cryptococcal meningitis. The NIAID Mycoses Study Group and the AIDS Clinical Trials Group. N Engl J Med 1992;326:83--9.
- Bicanic T, Harrison T, Niepieklo A, et al. Symptomatic relapse of HIV-associated cryptococcal meningitis after initial fluconazole monotherapy: the role of fluconazole resistance and immune reconstitution. Clin Infect Dis 2006;43:1069--73.
- Bicanic T, Meintjes G, Wood R, et al. Fungal burden, early fungicidal activity, and outcome in cryptococcal meningitis in antiretroviral-naive or antiretroviral-experienced patients treated with amphotericin B or fluconazole. Clin Infect Dis 2007;45:76--80.
- Saag MS, Cloud GA, Graybill JR, et al. A comparison of itraconazole versus fluconazole as maintenance therapy for AIDS-associated cryptococcal meningitis. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Clin Infect Dis 1999;28:291--6.
- Graybill JR, Sobel J, Saag M, et al. Diagnosis and management of increased intracranial pressure in patients with AIDS and cryptococcal meningitis. The NIAID Mycoses Study Group and AIDS Cooperative Treatment Groups. Clin Infect Dis 2000;30:47--54.
- Newton PN, Thai le H, Tip NQ, et al. A randomized, double-blind, placebo-controlled trial of acetazolamide for the treatment of elevated intracranial pressure in cryptococcal meningitis. Clin Infect Dis 2002;35:769--72.
- Powderly WG, Cloud GA, Dismukes WE, et al. Measurement of cryptococcal antigen in serum and cerebrospinal fluid: value in the management of AIDS-associated cryptococcal meningitis. Clin Infect Dis 1994;18:789--92.
- Aberg JA, Watson J, Segal M, et al. Clinical utility of monitoring serum cryptococcal antigen (sCRAG) titers in patients with AIDS-related cryptococcal disease. HIV Clin Trials 2000;1:1--6.
- Lortholary O, Fontanet A, Mémain N, et al. Incidence and risk factors of immune reconstitution inflammatory syndrome complicating HIV-associated cryptococcosis in France. AIDS 2005;19:1043--9.
- Skiest DJ, Hester LJ, Hardy RD. Cryptococcal immune reconstitution inflammatory syndrome: report of four cases in three patients and review of the literature. J Infect 2005;51:e289--97.
- Lesho E. Evidence base for using corticosteroids to treat HIV-associated immune reconstitution syndrome. Expert Rev Anti Infect Ther 2006;4:469--78.
- Lortholary O. Management of cryptococcal meningitis in AIDS: the need for specific studies in developing countries. Clin Infect Dis 2007;45:81--3.
- Bozzette SA, Larsen RA, Chiu J, et al. A placebo-controlled trial of maintenance therapy with fluconazole after treatment of cryptococcal meningitis in the acquired immunodeficiency syndrome. California Collaborative Treatment Group. N Engl J Med 1991;324:580--4.
- Powderly WG, Saag MS, Cloud GA, et al. A controlled trial of fluconazole or amphotericin B to prevent relapse of cryptococcal meningitis in patients with the acquired immunodeficiency syndrome. The NIAID AIDS Clinical Trials Group and Mycoses Study Group. N Engl J Med 1992;326:793--8.
- Kirk O, Reiss P, Uberti-Foppa C, et al. Safe interruption of maintenance therapy against previous infection with four common HIV-associated opportunistic pathogens during potent antiretroviral therapy. Ann Intern Med 2002;137:239--50.
- Vibhagool A, Sungkanuparph S, Mootsikapun P, et al. Discontinuation of secondary prophylaxis for cryptococcal meningitis in human immunodeficiency virus-infected patients treated with highly active antiretroviral therapy: a prospective, multicenter, randomized study. Clin Infect Dis 2003;36:1329--31.
- Mussini C, Pezzotti P, Miro JM, et al. Discontinuation of maintenance therapy for cryptococcal meningitis in patients with AIDS treated with highly active antiretroviral therapy: an international observational study. Clin Infect Dis 2004;38:565--71.
- Whitt SP, Koch GA, Fender B, et al. Histoplasmosis in pregnancy: case series and report of transplacental transmission. Arch Intern Med 2004;164:454--8.
- Wheat LJ, Chetchotisakd P, Williams B, et al. Factors associated with severe manifestations of histoplasmosis in AIDS. Clin Infect Dis 2000;30:877--81.
- Schutze GE, Tucker NC, Jacobs RF. Histoplasmosis and perinatal human immunodeficiency virus. Pediatr Infect Dis J 1992;11:501--2.
- Wheat LJ, Freifeld AG, Kleiman MB, et al. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007;45:807--25.
- Wheat LJ, Musial CE, Jenny-Avital E. Diagnosis and management of central nervous system histoplasmosis. Clin Infect Dis 2005;40:844--52.
- Saidinejad M, Burns MM, Harper MB. Disseminated histoplasmosis in a nonendemic area. Pediatr Infect Dis J 2004;23:781--2.
- Byers M, Feldman S, Edwards J. Disseminated histoplasmosis as the acquired immunodeficiency syndrome-defining illness in an infant. Pediatr Infect Dis J 1992;11:127--8.
- Pillay T, Pillay DG, Bramdev A. Disseminated histoplasmosis in a human immunodeficiency virus-infected African child. Pediatr Infect Dis J 1997;16:417--8.
- Odio CM, Navarrete M, Carrillo JM, et al. Disseminated histoplasmosis in infants. Pediatr Infect Dis J 1999;18:1065--8.
- Leggiadro RJ, Barrett FF, Hughes WT. Disseminated histoplasmosis of infancy. Pediatr Infect Dis J 1988;7:799--805.
- Hughes WT. Hematogenous histoplasmosis in the immunocompromised child. J Pediatr 1984;105:569--75.
- Wheat LJ. Antigen detection, serology, and molecular diagnosis of invasive mycoses in the immunocompromised host. Transpl Infect Dis 2006;8:128--39.
- Wheat LJ. Improvements in diagnosis of histoplasmosis. Expert Opin Biol Ther 2006;6:1207--21.
- Brandt ME, Warnock DW. Histoplasma, Blastomyces, Coccidioides, and other dimorphic fungi causing systemic mycoses. In Manual of clinical microbiology, 9th ed. P. Murray, ed. 2007;9:1857--65.
- Bialek R, Feucht A, Aepinus C, et al. Evaluation of two nested PCR assays for detection of Histoplasma capsulatum DNA in human tissue. J Clin Microbiol 2002;40:1644--7.
- Tang YM, Li H, Durkin MM, et al. Urine polymerase chain reaction is not as sensitive as urine antigen for the diagnosis of disseminated histoplasmosis. Diagn Microbiol Infect Dis 2006;54:283--7.
- Tobon AM, Agudelo CA, Rosero DS, et al. Disseminated histoplasmosis: a comparative study between patients with acquired immunodeficiency syndrome and non-human immunodeficiency virus-infected individuals. Am J Trop Med Hyg 2005;73:576--82.
- Fojtasek MF, Kleiman MB, Connolly-Stringfield P, et al. The Histoplasma capsulatum antigen assay in disseminated histoplasmosis in children. Pediatr Infect Dis J 1994;13:801--5.
- Lenhart SW, Schafer MP, Singal M, et al. Histoplasmosis---protecting workers at risk. Atlanta, GA: U.S. Department of Health and Human Services, CDC; 2004. DHHS (NIOSH) Publication No. 2005-109. Available at http://www.cdc.gov/niosh/docs/2005-109/#a.
- Goldman M, Zackin R, Fichtenbaum CJ, et al. Safety of discontinuation of maintenance therapy for disseminated histoplasmosis after immunologic response to antiretroviral therapy. Clin Infect Dis 2004;38:1485--9.
- Dismukes WE, Bradsher RW, Cloud GC, et al. Itraconazole therapy for blastomycosis and histoplasmosis. NIAID Mycoses Study Group. Am J Med 1992;93:489--97.
- Wheat J, Hafner R, Korzun AH, et al. Itraconazole treatment of disseminated histoplasmosis in patients with the acquired immunodeficiency syndrome. AIDS Clinical Trial Group. Am J Med 1995;98:336--42.
- Wheat LJ, Connolly P, Smedema M, et al. Emergence of resistance to fluconazole as a cause of failure during treatment of histoplasmosis in patients with acquired immunodeficiency disease syndrome. Clin Infect Dis 2001;33:1910--3.
- Adderson EE. Histoplasmosis in a pediatric oncology center. J Pediatr 2004;144:100--6.
- Wheat LJ, Connolly-Stringfield P, Blair R, et al. Effect of successful treatment with amphotericin B on Histoplasma capsulatum variety capsulatum polysaccharide antigen levels in patients with AIDS and histoplasmosis. Am J Med 1992;92:153--60.
- Nacher M, Sarazin F, El Guedj M, et al. Increased incidence of disseminated histoplasmosis following highly active antiretroviral therapy initiation. J Acquir Immune Defic Syndr 2006;41:468--70.
- Vargas SL, Hughes WT, Santolaya ME, et al. Search for primary infection by Pneumocystis carinii in a cohort of normal, healthy infants. Clin Infect Dis 2001;32:855--61.
- Respaldiza N, Medrano FJ, Medrano AC, et al. High seroprevalence of Pneumocystis infection in Spanish children. Clin Microbiol Infect 2004;10:1029--31.
- Pifer LL, Hughes WT, Stagno S, et al. Pneumocystis carinii infection: evidence for high prevalence in normal and immunosuppressed children. Pediatrics 1978;61:35--41.
- Simonds RJ, Oxtoby MJ, Caldwell MB, et al. Pneumocystis carinii pneumonia among US children with perinatally acquired HIV infection. JAMA 1993;270:470--3.
- Gibb DM, Davison CF, Holland FJ, et al. Pneumocystis carinii pneumonia in vertically acquired HIV infection in the British Isles. Arch Dis Child 1994;70:241--4.
- Williams AJ, Duong T, McNally LM, et al. Pneumocystis carinii pneumonia and cytomegalovirus infection in children with vertically acquired HIV infection. AIDS 2001;15:335--9.
- Morris A, Lundgren JD, Masur H, et al. Current epidemiology of Pneumocystis pneumonia. Emerg Infect Dis 2004;10:1713--20.
- Ikeogu MO, Wolf B, Mathe S. Pulmonary manifestations in HIV seropositivity and malnutrition in Zimbabwe. Arch Dis Child 1997;76:124--8.
- Chintu C, Mudenda V, Lucas S, et al. Lung diseases at necropsy in African children dying from respiratory illnesses: a descriptive necropsy study. Lancet 2002;360:985--90.
- Madhi SA, Cutland C, Ismail K, et al. Ineffectiveness of trimethoprim--sulfamethoxazole prophylaxis and the importance of bacterial and viral coinfections in African children with Pneumocystis carinii pneumonia. Clin Infect Dis 2002;35:1120--6.
- Hendley JO, Weller TH. Activation and transmission in rats of infection with Pneumocystis. Proc Soc Exp Biol Med 1971;137:1401--4.
- Hughes WT. Natural mode of acquisition for de novo infection with Pneumocystis carinii. J Infect Dis 1982;145:842--8.
- de Boer MG, Bruijnesteijn van Coppenraet LE, Gaasbeek A, et al. An outbreak of Pneumocystis jiroveci pneumonia with 1 predominant genotype among renal transplant recipients: interhuman transmission or a common environmental source? Clin Infect Dis 2007;44:1143--9.
- Hocker B, Wendt C, Nahimana A, et al. Molecular evidence of Pneumocystis transmission in pediatric transplant unit. Emerg Infect Dis 2005;11:330--2.
- Rabodonirina M, Vanhems P, Couray-Targe S, et al. Molecular evidence of interhuman transmission of Pneumocystis pneumonia among renal transplant recipients hospitalized with HIV-infected patients. Emerg Infect Dis 2004;10:1766--73.
- Mortier E, Pouchot J, Bossi P, et al. Maternal-fetal transmission of Pneumocystis carinii in human immunodeficiency virus infection. N Engl J Med 1995;332:825.
- Fatti GL, Zar HJ, Swingier GH. Clinical indicators of Pneumocystis jiroveci pneumonia (PCP) in South African children infected with the human immunodeficiency virus. Int J Infect Dis 2006;10:282--5.
- Podlekareva D, Mocroft A, Dragsted UB, et al. Factors associated with the development of opportunistic infections in HIV-1-infected adults with high CD4+ cell counts: a EuroSIDA study. J Infect Dis 2006;194:633--41.
- Ng VI, Yajko DM, Hadley WK. Extrapulmonary pneumocystosis. Clin Microbiol Rev 1997;10:401--18.
- Chen A, Zaidi AK, Mueller BU, et al. Pneumocystis carinii presenting as a mediastinal mass in a child with acquired immunodeficiency syndrome. Pediatr Infect Dis J 1999;18:827--31.
- Surve TY, Rathod AD. Role of nasogastric aspirate in HIV-positve children presenting with respiratory symptoms. J Trop Pediatr 2006;52:451--3.
- Chan H, Pifer L, Hughes WT, et al. Comparison of gastric contents to pulmonary aspirates for the cytologic diagnosis of Pneuomcystis carinii pneumonia. J Pediatr 1977;90:243--4.
- Huang L, Morris A, Limper AH, et al. An Official ATS Workshop Summary: Recent advances and future directions in Pneumocystis pneumonia (PCP). Proc Am Thorac Soc 2006;3:655--64.
- Lebovitz E, Pollack H, Rigaud M, et al. Polymerase chain reaction is more sensitive than standard cytologic stains in detecting Pneumocystis carinii in bronchoalveolar lavages from human immunodeficiency virus type 1-infected infants and children with pneumonia. Pediatr Infect Dis J 1995;14:714--6.
- Maskell NA, Waine DJ, Lindley A, et al. Asymptomatic carriage of Pneumocystis jiroveci in subjects undergoing bronchoscopy: a prospective study. Thorax 2003;58:594--7.
- Glatman-Freedman A, Ewig JM, Dobroszycki J, et al. Simultaneous Pneumocystis carinii and pneumococcal pneumonia in human immunodeficiency virus-infected children. J Pediatr 1998;132:169--71.
- Jeena PM, Coovadia HM, Chrystal V. Pneumocystis carinii and cytomegalovirus infections in severely ill, HIV-infected African infants. Ann Trop Paediatr 1996;16:361--8.
- CDC. Guidelines for prophylaxis against Pneumocystis carinii pneumonia for children infected with human immunodeficiency virus. MMWR 1991;40(No. RR-2).
- Hughes WT, Rivera GK, Schell MJ, et al. Successful intermittent chemoprophylaxis for Pneumocystis carinii pneumonitis. N Engl J Med 1987;316:1627--32.
- Hughes WT, Kuhn S, Chaudhary S, et al. Successful chemoprophylaxis for Pneumocystis carinii pneumonitis. N Engl J Med 1977;297:1419--26.
- Carr A, Tindall B, Brew BJ, et al. Low-dose trimethoprim--sulfamethoxazole prophylaxis for toxoplasmic encephalitis in patients with AIDS. Ann Intern Med 1992;117:106--11.
- Bozzette SA, Finkelstein DM, Spector SA, et al. A randomized trial of three antipneumocystis agents in patients with advanced human immunodeficiency virus infection. NIAID AIDS Clinical Trials Group. N Engl J Med 1995;332:693--9.
- Hardy WD, Feinberg J, Finkelstein DM, et al. A controlled trial of trimethoprim--sulfamethoxazole or aerosolized pentamidine for secondary prophylaxis of Pneumocystis carinii pneumonia in patients with the acquired immunodeficiency syndrome. AIDS Clinical Trials Group Protocol 021. N Engl J Med 1992;327:1842--8.
- Dworkin MS, Williamson J, Jones JL, et al. Prophylaxis with trimethoprim--sulfamethoxazole for human immunodeficiency virus-infected patients: impact on risk for infectious diseases. Clin Infect Dis 2001;33:393--8.
- Martin JN, Rose DA, Hadley WK, et al. Emergence of trimethoprim--sulfamethoxazole resistance in the AIDS era. J Infect Dis 1999;180:1809--18.
- Huovinen P. Resistance to trimethoprim--sulfamethoxazole. Clin Infect Dis 2001;32:1608--14.
- El-Sadr WM, Murphy RL, Yurik TM, et al. Atovaquone compared with dapsone for the prevention of Pneumocystis carinii pneumonia in patients with HIV infection who cannot tolerate trimethoprim, sulfonamides, or both. Community Program for Clinical Research on AIDS and the AIDS Clinical Trials Group. N Engl J Med 1998;339:1889--95.
- Hughes WT. Use of dapsone in the prevention and treatment of Pneumocystis carinii pneumonia: a review. Clin Infect Dis 1998;27:191--204.
- Principi N, Marchisio P, Onorato J, et al. Long-term administration of aerosolized pentamidine as primary prophylaxis against Pneumocystis carinii pneumonia in infants and children with symptomatic human immunodeficiency virus infection. The Italian Pediatric Collaborative Study Group on Pentamidine. J Acquir Immune Defic Syndr Hum Retrovirol 1996;12:158--63.
- Milstone AM, Balakrishnan SL, Foster CB, et al. Failure of intravenous pentamidine prophylaxis to prevent Pneumocystis pneumonia in a pediatric hematopoietic stem cell transplant (HSCT) patient. Pediatr Blood Cancer 2006;47:859--60.
- Furrer H, Egger M, Opravil M, et al. Discontinuation of primary prophylaxis against Pneumocystis carinii pneumonia in HIV-1-infected adults treated with combination antiretroviral therapy. Swiss HIV Cohort Study. N Engl J Med 1999;340:1301--6.
- Schneider MM, Borleffs JC, Stolk RP, et al. Discontinuation of prophylaxis for Pneumocystis carinii pneumonia in HIV-1-infected patients treated with highly active antiretroviral therapy. Lancet 1999;353:201--3.
- Dworkin MS, Hanson DL, Kaplan JE, et al. Risk for preventable opportunistic infections in persons with AIDS after antiretroviral therapy increases CD4+ T lymphocyte counts above prophylaxis thresholds. J Infect Dis 2000;182:611--5.
- Ledergerber B, Mocroft A, Reiss P, et al. Discontinuation of secondary prophylaxis against Pneumocystis carinii pneumonia in patients with HIV infection who have a response to antiretroviral therapy. Eight European Study Groups. N Engl J Med 2001;344:168--74.
- Lopez Bernaldo de Quiros JC, Miro JM, Pena JM, et al. A randomized trial of the discontinuation of primary and secondary prophylaxis against Pneumocystis carinii pneumonia after highly active antiretroviral therapy in patients with HIV infection. Grupo de Estudio del SIDA 04/98. N Engl J Med 2001;344:159--67.
- Zar JH, Langdon G, Apolles P, et al. Oral trimethoprim--sulphamethoxazole levels in stable HIV-infected children. S Afr Med J 2006;96:627--9.
- Leoung GS, Mills J, Hopewell PC, et al. Dapsone-trimethoprim for Pneumocystis carinii pneumonia in the acquired immunodeficiency syndrome. Ann Intern Med 1986;105:45--8.
- Mirochnick M, Cooper E, McIntosh K, et al. Pharmacokinetics of dapsone administered daily and weekly in human immunodeficiency virus-infected children. Antimicrob Agents Chemother 1999;43:2586--91.
- Mills J, Leoung G, Medina I, et al. Dapsone treatment of Pneumocystis carinii pneumonia in the acquired immunodeficiency syndrome. Antimicrob Agents Chemother 1988;32:1057--60.
- Sleasman JW, Hemenway C, Klein AS, et al. Corticosteroids improve survival of children with AIDS and Pneumocystis carinii pneumonia. Am J Dis Child 1993;147:30--4.
- Bye MR, Cairns-Bazarian AM, Ewig JM. Markedly reduced mortality associated with corticosteroid therapy of Pneumocystis carinii pneumonia in children with acquired immunodeficiency syndrome. Arch Pediatr Adolesc Med 1994;148:638--41.
- McLaughlin GE, Virdee SS, Schleien CL, et al. Effect of corticosteroids on survival of children with acquired immunodeficiency syndrome and Pneumocystis carinii-related respiratory failure. J Pediatr 1995;126(5 Pt 1):821--4.
- Creery WE, Hashmi A, Hutchison JS, et al. Surfactant therapy improves pulmonary function in infants with Pneumocystis carinii pneumonia and acquired immunodeficiency syndrome. Pediatr Pulmonol 1997;24:370--3.
- Marriage S, Underhill H, Nadel S. Use of natural surfactant in an HIV-infected infant with Pneumocystis carinii pneumonia. Intensive Care Med 1995;22:611--2.
- Slater A, Nichani SH, Macrae D, et al. Surfactant adjunctive therapy for Pneumocystis carinii pneumonitis in an infant with acute lymphoblastic leukaemia. Intensive Care Med 1995;21:261--3.
- Schmidt R, Markart P, Ruppert C, et al. Pulmonary surfactant in patients with Pneumocystis pneumonia and acquired immunodeficiency syndrome. Crit Care med 2006;34:2370--6.
- Datta D, Ali SA, Henken EM, et al. Pneumocystis carinii pneumonia: the time course of clinical and radiographic improvement. Chest 2003;124:1820--3.
- Ratnam I, Chiu C, Kandala NB, et al. Incidence and risk factors for immune reconstitution inflammatory syndrome in an ethnically diverse HIV type 1-infected cohort. Clin Infect Dis 2006;42:418--27.
- Gutman LT. The use of trimethoprim--sulfamethoxazole in children: a review of adverse reactions and indications. Pediatr Infect Dis J 1984;3:349--57.
- Rieder MJ, King SM, Read S. Adverse reactions to trimethoprim--sulfamethoxazole among children with human immunodeficiency virus infection. Pediatr Infect Dis J 1997;16:1028--31.
- Para MF, Finkelstein D, Becker S, et al. Reduced toxicity with gradual initiation of trimethoprim--sulfamethoxazole as primary prophylaxis for Pneumocystis carinii pneumonia: AIDS Clinical Trials Group 268. J Acquir Immune Defic Syndr 2000;24:337--43.
- Leoung GS, Stanford JF, Giordano MF, et al. Trimethoprim--sulfamethoxazole (TMP-SMZ) dose escalation versus direct rechallenge for Pneumocystis carinii pneumonia prophylaxis in human immunodeficiency virus-infected patients with previous adverse reaction to TMP-SMZ. J Infect Dis 2001;184:992--7.
- Goodwin SD. Pneumocystis carinii pneumonia in human immunodeficiency virus-infected infants and children. Pharmacotherapy 1993;13:640--6.
- Mussini C, Pezzotti P, Antinori A, et al. Discontinuation of secondary prophylaxis for Pneumocystis carinii pneumonia in human immunodeficiency virus-infected patients: a randomized trial by the CIOP Study Group. Clin Infect Dis 2003;36:645--51.
- Leav BA, Mackay M, Ward HD. Cryptosporidium species: new insights and old challenges Clin Infect Dis 2003;36:903--8.
- Heijbel H, Slaine K, Seigel B, et al. Outbreak of diarrhea in a day care center with spread to household members: the role of Cryptosporidium. Pediatr Infect Dis J 1987;6:532--5.
- Puech MC, McAnulty JM, Lesjak M, et al. A statewide outbreak of cryptosporidiosis in New South Wales associated with swimming at public pools. Epidemiol Infect 2001;126:389--96.
- Mead PS, Slutsker L, Dietz V, et al. Food-related illness and death in the United States. Emerg Infect Dis 1999;5:607--25.
- Pettoello-Mantoani M, Di Martino L, Dettori G, et al. Asymptomatic carriage of intestinal Cryptosporidium in immunocompetent and immunodeficient children: a prospective study. Pediatr Infect Dis J 1995;14:1042--7.
- Cegielski JP, McKee S, Madden JF, et al. Cryptosporidium, Enterocytozoon, and Cyclospora infections in pediatric and adult patients with diarrhea in Tanzania. Clin Infect Dis 1999;28:314--21.
- Tumwine JK, Kekitiinwa A, Nabukeera N, et al. Cryptosporidium parvum in children with diarrhea in Mulago Hospital, Kampala, Uganda. Am J Trop Med Hyg 2003;68:710--5.
- Huang DB, White AC. An updated review on Cryptosporidium and Giardia. Gastroenterol Clin North Am 2006;35:291--314.
- Chen XM, Keithly JS, Paya CV, et al. Cryptosporidiosis. N Engl J Med 2002;346:1723--31.
- Mathis A. Microsporidia: emerging advances in understanding the basic biology of these unique organisms. Int J Parasitol 2000;30:795--804.
- Hutin YJ, Sombardier MN, Liguory O, et al. Risk factors for intestinal microsporidiosis in patients with human immunodeficiency virus infection: a case-control study. J Infect Dis 1998;178:904--7.
- Newman RD, Sears CL, Moore SR, et al. Longitudinal study of Cryptosporidium infection in children in northeastern Brazil. J Infect Dis 1999;180:167--75.
- Lima AA, Moore SR, Barboza MS Jr, et al. Persistent diarrhea signals a critical period of increased diarrhea burdens and nutritional shortfalls: a prospective cohort study among children in northeastern Brazil. J Infect Dis 2000;181:1643--51.
- Vakil NB, Schwartz SM, Buggy BP, et al. Biliary cryptosporidiosis in HIV-infected people after the waterborne outbreak of cryptosporidiosis in Milwaukee. N Engl J Med 1996;334:19--23.
- Kotler DP, Orenstein JM. Clinical syndromes associated with microsporidiosis. Adv Parasitol 1998: 40:321--49.
- Weber R, Bryan RT, Bishop HS, et al. Threshold of detection of Cryptosporidium oocysts in human stool specimens: evidence for low sensitivity of current diagnostic methods. J Clin Microbiol 1991;29:1323--7.
- Garcia LS, Shimizu RY. Evaluation of nine immunoassay kits (enzyme immunoassay and direct fluorescence) for detection of Giardia lamblia and Cryptosporidium parvum in human fecal specimens. J Clin Microbiol 1997;35:1526--9.
- Garcia LS, Shimizu RY, Novak S, et al. Commercial assay for detection of Giardia lamblia and Cryptosporidium parvum antigens in human fecal specimens by rapid solid-phase qualitative immunochromatography. J Clin Microbiol 2003;41:209--12.
- McLauchlin J, Amar CF, Pedraza-Díaz S, et al. Polymerase chain reaction-based diagnosis of infection with Cryptosporidium in children with primary immunodeficiencies. Pediatr Infect Dis J 2003;22:329--35.
- Menotti J, Cassinat B, Porcher R, et al. Development of a real-time polymerase-chain-reaction assay for quantitative detection of Enterocytozoon bieneusi DNA in stool specimens from immunocompromised patients with intestinal microsporidiosis. J Infect Dis 2003;187:1469--74.
- Holmberg SD, Moorman AC, Von Bargen JC, et al. Possible effectiveness of clarithromycin and rifabutin for cryptosporidiosis chemoprophylaxis in HIV disease. HIV Outpatient Study (HOPS) Investigators. JAMA 1998;279:384--6.
- Fichtenbaum CJ, Zackin R, Feinberg J, et al. Rifabutin but not clarithromycin prevents cryptosporidiosis in persons with advanced HIV infection. AIDS 2000;14:2889--93.
- Miao YM, Awad-El-Kariem FM, Franzen C, et al. Eradication of cryptosporidia and microsporidia following successful antiretroviral therapy. J Acquir Immune Defic Syndr 2000;25:124--9.
- Abubakar I, Abubakar I, Aliyu SH, et al. Prevention and treatment of cryptosporidiosis in immunocompromised patients. Cochrane Database Syst Rev 2007;Jan 24:CD004932.
- Rossignol JF, Ayoub A, Ayers MS. Treatment of diarrhea caused by Cryptosporidium parvum: a prospective randomized, double-blind, placebo-controlled study of Nitazoxanide. J Infect Dis 2001;184:103--6.
- Amadi B, Mwiya M, Musuku J, et al. Effect of nitazoxanide on morbidity and mortality in Zambian children with cryptosporidiosis: a randomised controlled trial Lancet 2000;360:1375--80.
- Rossignol JF, Hidalgo H, Feregrino M, et al. A double-"blind" placebo-controlled study of nitazoxanide in the treatment of cryptosporidial diarrhoea in AIDS patients in Mexico. Trans R Soc Trop Med Hyg 1998;92:663--6.
- White AC, Chappell CL, Hayat CS, et al. Paromomycin for cryptosporidiosis in AIDS: a prospective, double-blind trial. J Infect Dis 1994;170:419--24.
- Hewitt RG, Yiannoutsos CT, Higgs ES, et al. Paromomycin: no more effective than placebo for treatment of cryptosporidiosis in patients with advanced human immunodeficiency virus infection. AIDS Clinical Trial Group. Clin Infect Dis 2000;31:1084--92.
- Hicks P, Zwiener RJ, Squires J, et al. Azithromycin therapy for Cryptosporidium parvum infection in four children infected with human immunodeficiency virus. J Pediatr 1996;129:297--300.
- Weber R, Sauer B, Lüthy R, et al. Intestinal coinfection with Enterocytozoon bieneusi and Cryptosporidium in a human immunodeficiency virus-infected child with chronic diarrhea. Clin Infect Dis 1993;17:480--3.
- Molina JM, Chastang C, Goguel J, et al. Albendazole for treatment and prophylaxis of microsporidiosis due to Encephalitozoon intestinalis in patients with AIDS: a randomized double-blind controlled trial. J Infect Dis 1998;177:1373--7.
- Didier PJ, Phillips JN, Kuebler DJ, et al. Antimicrosporidial activities of fumagillin, TNP-470, ovalicin, and ovalicin derivatives in vitro and in vivo. Antimicrob Agents Chemother 2006;50:2146--55.
- Bicart-See A, Massip P, Linas MD, et al. Successful treatment with nitazoxanide of Enterocytozoon bieneusi microsporidiosis in a patient with AIDS. Antimicrob Agents Chemother 2000;44:167--8.
- Diesenhouse MC, Wilson LA, Corrent GF, et al. Treatment of microsporidial keratoconjunctivitis with topical fumagillin. Am J Ophthalmol 1993;115:293--8.
- Didier ES. Effects of albendazole, fumagillin, and TNP-470 on microsporidial replication in vitro. Antimicrob Agents Chemother 1997;41:1541--6.
- Thwing J, Skarbinski J, Newman RD, et al. Malaria surveillance---United States, 2005. In: Surveillance Summaries, June 8, 2007. MMWR 2007;56(No. SS-6):23--40.
- Rivera-Matos IR, Atkins JT, Doerr CA, et al. Pediatric malaria in Houston, Texas. Am J Trop Med Hyg 1997;57:560--3.
- Emanuel B, Aronson N, Shulman S. Malaria in children in Chicago. Pediatrics 1993;92:83--5.
- Miller KK, Banerji A. Epidemiology of malaria presenting at British Columbia's Children's Hospital, 1984--2001: lessons for prevention. Can J Public Health 2004;95:245--8.
- Whitworth J, Morgan D, Quigley M, et al. Effect of HIV-1 and increasing immunosuppression on malaria parasitaemia and clinical episodes in adults in rural Uganda: a cohort study. Lancet 2000;356:1051--6.
- Patnaik P, Jere CS, Miller WC, et al. Effects of HIV-1 serostatus, HIV-1 RNA concentration, and CD4 cell count on the incidence of malaria infection in a cohort of adults in rural Malawi. J Infect Dis 2005;192:984--91.
- Greenberg AE, Nsa W, Ryder RW, et al. Plasmodium falciparum malaria and perinatally acquired human immunodeficiency virus type 1 infection in Kinshasa, Zaire. A prospective, longitudinal cohort study of 587 children. N Engl J Med 1991;325:105--9.
- Taha TE, Canner JK, Dallabetta GA, et al. Childhood malaria parasitaemia and human immunodeficiency virus infection in Malawi. Trans R Soc Trop Med Hyg 1994;88:164--5.
- Kalyesubula I, Musoke-Mudido P, Marum L, et al. Effects of malaria infection in human immunodeficiency virus type 1-infected Ugandan children. Pediatr Infect Dis J 1997;16:876--81.
- Villamor E, Fataki MR, Mbise RL, et al. Malaria parasitaemia in relation to HIV status and vitamin A supplementation among pre-school children. Trop Med Int Health 2003;8:1051--61.
- Rogerson SR, Gladstone M, Callaghan M, et al. HIV infection among paediatric in-patients in Blantyre, Malawi. Trans R Soc Trop Med Hyg 2004;98:544--52.
- Grimwade K, French N, Mbatha DD, et al. Childhood malaria in a region of unstable transmission and high human immunodeficiency virus prevalence. Pediatr Infect Dis J 2003;22:1057--63.
- Otieno RO, Ouma C, Ong'echa JM, et al. Increased severe anemia in HIV-1-exposed and HIV-1-positive infants and children during acute malaria. AIDS 2006;20:275--80.
- Mermin J, Lule J, Ekwaru JP, et al. Effect of co-trimoxazole prophylaxis on morbidity, mortality, CD4-cell count, and viral load in HIV infection in rural Uganda. Lancet 2004;364:1428--34.
- Skarbinski J, James EM, Causer LM, et al. Malaria surveillance---United States, 2004. In: Surveillance Summaries, May 26, 2006. MMWR 2006;55(No. SS-4):23--37.
- Shingadia D, Shulman ST. Recognition and management of imported malaria in children Seminars in Pediatr Infect Dis 2000;11:172--7.
- Stauffer WM, Maroushek S, Kamat D. Medical screening of immigrant children. Clin Pediatr (Phila) 2003;42:763--73.
- Maroushek SR, Aguilar EF, Stauffer W, et al. Malaria among refugee children at arrival in the United States. Pediatr Infect Dis J 2005;24:450--2.
- Ndao M, Bandyayera E, Kokoskin E, et al. Comparison of blood smear, antigen detection, and nested-PCR methods for screening refugees from regions where malaria is endemic after a malaria outbreak in Quebec, Canada. J Clin Microbiol 2004;42:2694--700.
- Stauffer WM, Newberry AM, Cartwright CP, et al. Evaluation of malaria screening in newly arrived refugees to the United States by microscopy and rapid antigen capture enzyme assay. Pediatr Infect Dis J 2006;25:948--50.
- Bacaner N, Stauffer B, Boulware DR, et al. Travel medicine considerations for North American immigrants visiting friends and relatives. JAMA 2004;291:2856--64.
- Hill DR, Ericsson CD, Pearson RD, et al. The practice of travel medicine: guidelines by the Infectious Diseases Society of America. Clin Infect Dis 2006;43:1499--539.
- Stauffer WM, Kamat D, Magill AJ. Traveling with infants and children. Part IV: insect avoidance and malaria prevention. J Travel Med 2003;10:225--40.
- Kamya MR, Gasasira AF, Achan J, et al. Effects of trimethoprim--sulfamethoxazole and insecticide-treated bednets on malaria among HIV-infected Ugandan children. AIDS 2007;21:2059--66.
- Kamya MR, Gasasira AF, Yeka A, et al. Effect of HIV-1 infection on antimalarial treatment outcomes in Uganda: a population-based study. J Infect Dis 2006;193:9--15.
- Byakika-Kibwika P, Ddumba E, Kamya M. Effect of HIV-1 infection on malaria treatment outcome in Ugandan patients. Afr Health Sci 2007;7:86--92.
- Stauffer WM, Fischer PR. Diagnosis and treatment of malaria in children. Clin Infect Dis 2003;37:1340--8.
- CDC. New medication for severe malaria available under an investigational new drug protocol. MMWR 2007;56:769--770.
- Newton PN, McGready R, Fernandez F, et al. Manslaughter by fake artesunate in Asia---will Africa be next? PLoS Med 2006;3:e197.
- Guerina NG, Hsu HW, Meissner HC, et al. Neonatal serologic screening and early treatment for congenital Toxoplasma gondii infection. The New England Regional Toxoplasma Working Group. N Engl J Med 1994;330:1858--63.
- Jara M, Hsu HW, Eaton RB, et al. Epidemiology of congenital toxoplasmosis identified by population-based newborn screening in Massachusetts. Pediatr Infect Dis J 2001;20:1132--5.
- Smith KL, Wilson M, Hightower AW, et al. Prevalence of Toxoplasma gondii antibodies in US military recruits in 1989: comparison with data published in 1965. Clin Infect Dis 1996;23:1182--3.
- Jones JL, Kruszon-Moran D, Wilson M, et al. Toxoplasma gondii infection in the United States: seroprevalence and risk factors. Am J Epidemiol 2001;154:357--65.
- Dunn D, Wallon M, Peyron F, et al. Mother-to-child transmission of toxoplasmosis: risk estimates for clinical counselling Lancet 1999;353:1829--33.
- Montoya JG. Laboratory diagnosis of Toxoplasma gondii infection and toxoplasmosis. J Infect Dis 2002;185(Suppl 1):S73--82.
- Falusi O, French AL, Seaberg EC, et al. Prevalence and predictors of Toxoplasma seropositivity in women with and at risk for human immunodeficiency virus infection. Clin Infect Dis 2002;35:1414--7.
- Minkoff H, Remington JS, Holman S, et al. Vertical transmission of Toxoplasma by human immunodeficiency virus-infected women. Am J Obstet Gynecol 1997;176:555--9.
- Dunn D, Newell ML, Gilbert R. Low risk of congenital toxoplasmosis in children born to women infected with human immunodeficiency virus. Pediatr Infect Dis J 1997;16:84.
- Dunn DT, Newell ML, Gilbert R, et al. Low incidence of congenital toxoplasmosis in children born to women infected with human immunodeficiency virus. European Collaborative Study and Research Network on Congenital Toxoplasmosis. Eur J Obstet Gynecol Reprod Biol 1996;68:93--6.
- Mitchell CD, Erlich SS, Mastrucci MT, et al. Congenital toxoplasmosis occurring in infants perinatally infected with human immunodeficiency virus 1. Pediatr Infect Dis J 1990;9:512--8.
- D'Offizi G, Topino S, Anzidei G, et al. Primary Toxoplasma gondii infection in a pregnant human immunodeficiency virus-infected woman. Pediatr Infect Dis J 2002;21:981--2.
- Vogel N, Kirisits M, Michael E, et al. Congenital toxoplasmosis transmitted from an immunologically competent mother infected before conception. Clin Infect Dis 1996;23:1055--60.
- CDC. HIV/AIDS surveillance report. 1996:8(2):1--18.
- Sobanjo A, Ferguson DJ, Gross U. Primary acquired toxoplasmosis in a five-year-old child with perinatal human immunodeficiency virus type 1 infection. Pediatr Infect Dis J 1999;18:476--8.
- Wahn V, Kramer HH, Voit T, et al. Horizontal transmission of HIV infection between two siblings. Lancet 1986;2:694.
- King SM, Matlow A, Al-Hajjar S, et al. Toxoplasmic encephalitis in a child with HIV infection---United States. Pediatr AIDS and HIV Infect. Fetus to Adolesc 1992: 3:242--4.
- McAuley J, Boyer KM, Patel D, et al. Early and longitudinal evaluations of treated infants and children and untreated historical patients with congenital toxoplasmosis: the Chicago Collaborative Treatment Trial. Clin Infect Dis 1994;18:38--72.
- Medlock MD, Tilleli JT, Pearl GS. Congenital cardiac toxoplasmosis in a newborn with acquired immunodeficiency syndrome. Pediatr Infect Dis J 1990;9:129--32.
- Pinon JM, Dumon H, Chemla C, et al. Strategy for diagnosis of congenital toxoplasmosis: evaluation of methods comparing mothers and newborns and standard methods for postnatal detection of immunoglobulin G, M, and A antibodies. J Clin Microbiol 2001;39:2267--71.
- Wilson M, Jones JL, McAuley JB. Toxoplasma. In: Murray PR, Baron EJ, Jorgensen JH, Landry ML, Pfaller MA, eds. Manual of clinical microbiology, 9th ed. St. Louis, MO: ASM Press; 2007:2070--81.
- Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet 2004;363:1965--76.
- Wong SY, Hajdu MP, Ramirez R, et al. Role of specific immunoglobulin E in diagnosis of acute Toxoplasma infection and toxoplasmosis. J Clin Microbiol 1993;31:2952--9.
- Portegies P, Solod L, Cinque P, et al. Guidelines for the diagnosis and management of neurological complications of HIV infection. Eur J Neurol 2004;11:297--304.
- Offiah CE, Turnbull IW. The imaging appearances of intracranial CNS infections in adult HIV and AIDS patients. Clin Radiol 2006;61:393--401.
- U.S. Department of Agriculture. FoodSafety.gov: gateway to government food safety information. Washington, DC: US Departent of Agriculture, 2002. Available at http://www.foodsafety.gov.
- Podzamczer D, Salazar A, Jimenez J, et al. Intermittent trimethoprim--sulfamethoxazole compared with dapsone-pyrimethamine for the simultaneous primary prophylaxis of Pneumocystis pneumonia and toxoplasmosis in patients infected with HIV. Ann Intern Med 1995;122:755--61.
- Opravil M, Hirschel B, Lazzarin A, et al. Once-weekly administration of dapsone/pyrimethamine vs. aerosolized pentamidine as combined prophylaxis for Pneumocystis carinii pneumonia and toxoplasmic encephalitis in human immunodeficiency virus-infected patients. Clin Infect Dis 1995;20:531--41.
- Kirk O, Lundgren JD, Pedersen C, et al. Can chemoprophylaxis against opportunistic infections be discontinued after an increase in CD4 cells induced by highly active antiretroviral therapy? AIDS 1999;13:1647--51.
- Furrer H, Opravil M, Bernasconi E, et al. Stopping primary prophylaxis in HIV-1-infected patients at high risk of Toxoplasma encephalitis. Swiss HIV Cohort Study. Lancet 2000;355:2217--8.
- Mussini C, Pezzotti P, Govoni A, et al. Discontinuation of primary prophylaxis for Pneumocystis carinii pneumonia and toxoplasmic encephalitis in human immunodeficiency virus type I-infected patients: the changes in opportunistic prophylaxis study. J Infect Dis 2000;181:1635--42.
- Miro JM, Lopez JC, Podzamczer D, et al. Discontinuation of primary and secondary Toxoplasma gondii prophylaxis is safe in HIV-infected patients after immunological restoration with highly active antiretroviral therapy: results of an open, randomized, multicenter clinical trial. Clin Infect Dis 2006;43:79--89.
- SYROCOT (Systematic Review on Congenital Toxoplasmosis) Study Group, Thiébaut R, Leproust S, et al. Effectiveness of prenatal treatment for congenital toxoplasmosis: a meta-analysis of individual patients' data. Lancet 2007;369:115--22.
- McLeod R, Boyer K, Karrison T, et al. Outcome of treatment for congenital toxoplasmosis, 1981--2004: the National Collaborative Chicago-Based Congenital Toxoplasmosis Study. Clin Infect Dis 2006;42:1383--94.
- Soheilian M, Sadoughi MM, Ghajarnia M, et al. Prospective randomized trial of trimethoprim--sulfamethoxazole versus pyrimethamine and sulfadiazine in the treatment of ocular toxoplasmosis. Ophthalmology 2005;112:1876--82.
- Lawn SD. Immune reconstitution disease associated with parasitic infections following initiation of antiretroviral therapy. Curr Opin Infect Dis 2007;20:482--8.
- Katlama C, De Wit S, O'Doherty E, et al. Pyrimethamine-clindamycin vs. pyrimethamine-sulfadiazine as acute and long-term therapy for toxoplasmic encephalitis in patients with AIDS. Clin Infect Dis 1996;22:268--75.
- Dannemann B, McCutchan JA, Israelski D, et al. Treatment of toxoplasmic encephalitis in patients with AIDS. A randomized trial comparing pyrimethamine plus clindamycin to pyrimethamine plus sulfadiazine. The California Collaborative Treatment Group. Ann Intern Med 1992;116:33--43.
- Duval X, Pajot O, Le Moing V, et al. Maintenance therapy with cotrimoxazole for toxoplasmic encephalitis in the era of highly active antiretroviral therapy. AIDS 2004;18:1342--4.
- Soriano V, Dona C, Rodriguez-Rosado R, et al. Discontinuation of secondary prophylaxis for opportunistic infections in HIV-infected patients receiving highly active antiretroviral therapy. AIDS 2000;14:383--6.
- Bertschy S, Opravil M, Cavassini M, et al. Discontinuation of maintenance therapy against Toxoplasma encephalitis in AIDS patients with sustained response to anti-retroviral therapy. Clin Microbiol Infect 2006;12:666--71.
- Stagno S, Reynolds DW, Pass RF, et al. Breast milk and the risk of cytomegalovirus infection. N Engl J Med 1980;302:1073--6.
- Pass RF, Hutto C, Reynolds DW, et al. Increased frequency of cytomegalovirus infection in children in group day care. Pediatrics 1984;74:121--6.
- Adler SP. Cytomegalovirus and child day care. Evidence for an increased infection rate among day-care workers. N Engl J Med 1989;321:1290--6.
- Pass RF. Epidemiology and transmission of cytomegalovirus infection. J Infect Dis 1985;152:243--8.
- Stagno S, Pass RF, Cloud G, et al. Primary cytomegalovirus infection in pregnancy. Incidence, transmission to fetus, and clinical outcome. JAMA 1986;256:1904--8.
- Yow MD, Williamson DW, Leeds LJ, et al. Epidemiologic characteristics of cytomegalovirus infection in mothers and their infants. Am J Obstet Gynecol 1998;158:1189--95.
- Mussi-Pinhata MM, Yamamoto AY, Figueiredo LT, et al. Congenital and perinatal cytomegalovirus infection in infants born to mothers infected with human immunodeficiency virus. J Pediatr 1998;132:285--90.
- Quinn TC, Piot P, McCormick JB, et al. Serologic and immunologic studies in patients with AIDS in North America and Africa. The potential role of infectious agents as cofactors in human immunodeficiency virus infection. JAMA 1987;257:2617--21.
- Prober CG, Enright AM. Congenital cytomegalovirus (CMV) infections: hats off to Alabama J Pediatr 2003;143:4--6.
- Revello MG, Zavattoni M, Furione M, et al. Diagnosis and outcome of preconceptional and periconceptional primary human cytomegalovirus infections. J Infect Dis 2002;186:553--7.
- Fowler KB, Stagno S, Pass RF. Maternal immunity and prevention of congenital cytomegalovirus infection. JAMA 2003;289:1008-11.
- Azam AZ, Vial Y, Fawer CL, et al. Prenatal diagnosis of congenital cytomegalovirus infection. Obstet Gynecol 2001;97:443--8.
- Boppana SB, Fowler KB, Britt WJ, et al. Symptomatic congenital cytomegalovirus infection in infants born to mothers with preexisting immunity to cytomegalovirus. Pediatrics 1999;104:55--60.
- Boppana SB, Rivera LB, Fowler KB, et al. Intrauterine transmission of cytomegalovirus to infants of women with preconceptional immunity. N Engl J Med 2001;344:1366--71.
- Mostad SB, Kreiss JK, Ryncarz A, et al. Cervical shedding of herpes simplex virus and cytomegalovirus throughout the menstrual cycle in women infected with human immunodeficiency virus type 1. Am J Obstet Gynecol 2000;183:948--55.
- Kitchen BJ, Engler HD, Gill VJ, et al. Cytomegalovirus infection in children with human immunodeficiency virus infection. Pediatr Infect Dis J 1997;16:358--63.
- Jabs DA, Van Natta ML, Holbrook JT, et al. Longitudinal study of the ocular complications of AIDS: 1. Ocular diagnoses at enrollment. Ophthalmology 2007;114:780--6.
- Chandwani S, Kaul A, Bebenroth D, et al. Cytomegalovirus infection in human immunodeficiency virus type 1-infected children. Pediatr Infect Dis J 1996;15:310--4.
- Boppana SB, Pass RF, Britt WJ, et al. Symptomatic congenital cytomegalovirus infection: neonatal morbidity and mortality. Pediatr Infect Dis J 1992;11:93--9.
- Fowler KB, Boppana SB. Congenital cytomegalovirus (CMV) infection and hearing deficit. J Clin Virol 2006;35:226--31.
- Doyle M, Atkins JT, Rivera-Matos IR. Congenital cytomegalovirus infection in infants infected with human immunodeficiency virus type 1. Pediatr Infect Dis J 1996;15:1102--6.
- Zaknun D, Zangerle R, Kapelari K, et al. Concurrent ganciclovir and foscarnet treatment for cytomegalovirus encephalitis and retinitis in an infant with acquired immunodeficiency syndrome: case report and review. Pediatr Infect Dis J 1997;16:807--11.
- Mueller BU, MacKay K, Cheshire LB, et al. Cytomegalovirus ureteritis as a cause of renal failure in a child infected with the human immunodeficiency virus. Clin Infect Dis 1995;20:1040--3.
- Olivero MT, Nelson RP Jr, Andrews T, et al. Cytomegalovirus sinus disease in a human immunodeficiency virus-infected child. Pediatr Infect Dis J 1995;14:629--31.
- Marriage SC, Booy R, Hermione Lyall EG, et al. Cytomegalovirus myelitis in a child infected with human immunodeficiency virus type 1. Pediatr Infect Dis J 1996;15:549--51.
- Kalayjian RC, Cohen ML BR, Flanigan TP. Cytomegalovirus ventriculoencephalitis in AIDS. A syndrome with distinct clinical and pathologic features. Medicine (Baltimore) 1993;72:67--77.
- Nigro G, Krzysztofiak A, Gattinara GC, et al. Rapid progression of HIV disease in children with cytomegalovirus DNAemia. AIDS 1996;10:1127--33.
- Whitley RJ, Cloud G, Gruber W, et al. Ganciclovir treatment of symptomatic congenital cytomegalovirus infection: results of a phase II study. National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. J Infect Dis 1997;175:1080--6.
- Kimberlin DW, Lin CY, Sánchez PJ, et al. Effect of ganciclovir therapy on hearing in symptomatic congenital cytomegalovirus disease involving the central nervous system: a randomized, controlled trial J Pediatr 2003;143:16--25.
- Oliver S, Cloug G, Sánchez P, et al. Effect of ganciclovir therapy on neurodevelopmental outcomes in symptomatic congenital cytomegalovirus infections involving the central nervous system: a randomized, controlled study [abstract 752908]. Society for Pediatric Research, San Francisco, California, April 29, 2006. The Woodlands, Texas: Society for Pediatric Research; 2006: Available at http://www.abstracts2view.com/pas. Citation: E-PAS2006:59:2540.2.
- Studies of Ocular Complications of AIDS Research, Group in collaboration with the AIDS Clinical Trials Group. Foscarnet-Ganciclovir Cytomegalovirus Retinitis Trial. 4. Visual outcomes. Ophthalmology 1994;101:1250--61.
- Musch DC, Martin DF, Gordon JF, et al. Treatment of cytomegalovirus retinitis with a sustained-release ganciclovir implant. The Ganciclovir Implant Study Group. N Engl J Med 1997;337:83--90.
- Martin DF, Sierra-Madero J, Walmsley S, et al. A controlled trial of valganciclovir as induction therapy for cytomegalovirus retinitis. N Engl J Med 2002;346:1119--26.
- Kempen JH, Jabs DA, Wilson LA, et al. Risk of vision loss in patients with cytomegalovirus retinitis and the acquired immunodeficiency syndrome. Arch Ophthalmol 2003;121:466--76.
- Studies of Ocular Complications of AIDS Research Group, The AIDS Clinical Trials Group. The ganciclovir implant plus oral ganciclovir versus parenteral cidofovir for the treatment of cytomegalovirus retinitis in patients with acquired immunodeficiency syndrome: the Ganciclovir Cidofovir Cytomegalovirus Retinitis Trial. Am J Ophthalmol 2001;131:457--67.
- Acosta EP, Brundage RC, King JR, et al. Ganciclovir population pharmacokinetics in neonates following intravenous administration of ganciclovir and oral administration of a liquid valganciclovir formulation. Clin Pharmacol Ther 2007;81:867--72.
- Walton RC, Whitcup SM, Mueller BU, et al. Combined intravenous ganciclovir and foscarnet for children with recurrent cytomegalovirus retinitis. Ophthalmology 1995;102:1865--70.
- Butler KM, De Smet MD, Husson RN, et al. Treatment of aggressive cytomegalovirus retinitis with ganciclovir in combination with foscarnet in a child infected with human immunodeficiency virus. J Pediatr 1992;120:483--6.
- Fletcher C, Sawchuk R, Chinnock B, et al. Human pharmacokinetics of the antiviral drug DHPG. Clin Pharmacol Ther 1986;40:281--6.
- Nguyen QD, Kempen JH, Bolton SG, et al. Immune recovery uveitis in patients with AIDS and cytomegalovirus retinitis after highly active antiretroviral therapy. Am J Ophthalmol 2000;129:634--9.
- Kosobucki BR, Goldberg DE, Bessho K, et al. Valganciclovir therapy for immune recovery uveitis complicated by macular edema. Am J Ophthalmol 2004;137:636--8.
- Martin DF, Kuppermann BD, Wolitz RA, et al. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. Roche Ganciclovir Study Group. N Engl J Med 1999;340:1063--70.
- Drew WL, Ives D, Lalezari JP, et al. Oral ganciclovir as maintenance treatment for cytomegalovirus retinitis in patients with AIDS. Syntex Cooperative Oral Ganciclovir Study Group. N Engl J Med 1995;333:615--20.
- Parenteral cidofovir for cytomegalovirus retinitis in patients with AIDS: the HPMPC peripheral cytomegalovirus retinitis trial. A randomized, controlled trial. Studies of Ocular complications of AIDS Research Group in Collaboration with the AIDS Clinical Trials Group. Ann Intern Med 1997;126:264--74.
- Palestine AG, Polis MA, De Smet MD, et al. A randomized, controlled trial of foscarnet in the treatment of cytomegalovirus retinitis in patients with AIDS. Ann Intern Med 1991;115:665--73.
- Spector SA, Weingeist T, Pollard RB, et al. A randomized, controlled study of intravenous ganciclovir therapy for cytomegalovirus peripheral retinitis in patients with AIDS. AIDS Clinical Trials Group and Cytomegalovirus Cooperative Study Group. J Infect Dis 1993;168:557--63.
- The Studies of the Ocular Complications of AIDS Research Group, Group icwtACT. Combination foscarnet and ganciclovir therapy vs monotherapy for the treatment of relapsed cytomegalovirus retinitis in patients with AIDS. The cytomegalovirus retreatment trial. Arch Ophthalmol 1996;114:23--33.
- Diaz-Llopis M, España E, Muñoz G, et al. High dose intravitreal foscarnet in the treatment of cytomegalovirus retinitis in AIDS. Br J Ophthalmol 1994;78:120--4.
- de Smet MD, Meenken CJ, van den Horn GJ. Fomivirsen---a phosphorothioate oligonucleotide for the treatment of CMV retinitis. Ocul Immunol Inflamm 1999;7:189--98.
- Kirsch LS, Arevalo JF, Chavez de la Paz E, et al. Intravitreal cidofovir (HPMPC) treatment of cytomegalovirus retinitis in patients with acquired immune deficiency syndrome. Ophthalmology 1995;102:533--42; discussion 542--3.
- Young S, Morlet N, Besen G, et al. High-dose (2000-microgram) intravitreous ganciclovir in the treatment of cytomegalovirus retinitis. Ophthalmology 1998;105:1404--10.
- Tural C, Romeu J, Sirera G, et al. Long-lasting remission of cytomegalovirus retinitis without maintenance therapy in human immunodeficiency virus-infected patients. J Infect Dis 1998;177:1080--3.
- Vrabec TR, Baldassano VF, Whitcup SM. Discontinuation of maintenance therapy in patients with quiescent cytomegalovirus retinitis and elevated CD4+ counts. Ophthalmology 1998;105:1259--64.
- Macdonald JC, Torriani FJ, Morse LS, et al. Lack of reactivation of cytomegalovirus (CMV) retinitis after stopping CMV maintenance therapy in AIDS patients with sustained elevations in CD4 T cells in response to highly active antiretroviral therapy. J Infect Dis 1998;177:1182--7.
- Whitcup SM, Fortin E, Lindblad AS, et al. Discontinuation of anticytomegalovirus therapy in patients with HIV infection and cytomegalovirus retinitis. JAMA 1999;282:1633--7.
- Jabs DA, Bolton SG, Dunn JP, et al. Discontinuing anticytomegalovirus therapy in patients with immune reconstitution after combination antiretroviral therapy. Am J Ophthalmol 1998;126:817--22.
- Jouan M, Saves M, Tubiana R, et al. Discontinuation of maintenance therapy for cytomegalovirus retinitis in HIV-infected patients receiving highly active antiretroviral therapy. AIDS 2001;15:23--31.
- Torriani FJ, Freeman WR, Macdonald JC, et al. CMV retinitis recurs after stopping treatment in virological and immunological failures of potent antiretroviral therapy. AIDS 2000;14:173--80.
- Lilleri D, Piccinini G, Genini E, et al. Monitoring of human cytomegalovirus (HCMV)-specific CD4+ T cell frequency by cytokine flow cytometry as a possible indicator for discontinuation of HCMV secondary prophylaxis in HAART-treated AIDS patients. J Clin Virol 2004;29:297--307.
- Tamarit A, Alberola J, Mira JV, et al. Assessment of human cytomegalovirus specific T cell immunity in human immunodeficiency virus infected patients in different disease stages following HAART and in long-term non-progressors. J Med Virol 2004;74:382--9.
- Weingberg A, Wiznia AA, Lafleur BJ, et al. Cytomegalovirus-specific cell-mediated immunity in HIV-infected children on HAART. AIDS Res Hum Retroviruses 2006;22:283--8.
- Saitoh A, Viani RM, Schrier RD, et al. Treatment of infants coinfected with HIV-1 and cytomegalovirus with combination antiretrovirals and ganciclovir. J Allergy Clin Immunol 2004;114:983--5.
- Spector SA, Wong R, Hsia K, et al. Plasma cytomegalovirus (CMV) DNA load predicts CMV disease and survival in AIDS patients. J Clin Invest 1998;101:497--502.
- Salmon-Ceron D, Mazeron MC, Chaput S, et al. Plasma cytomegalovirus DNA, pp65 antigenaemia and a low CD4 cell count remain risk factors for cytomegalovirus disease in patients receiving highly active antiretroviral therapy. AIDS 2000;14:1041--9.
- Chu CM, Karayiannis P, Fowler MJ, et al. Natural history of chronic hepatitis B virus infection in Taiwan: studies of hepatitis B virus DNA in serum. Hepatology 1985;5:431--4.
- Chang MH, Sung JL, Lee CY, et al. Factors affecting clearance of hepatitis B e antigen in hepatitis B surface antigen carrier children. J Pediatr 1989;115:385--90.
- Elisofon SA, Jonas MM. Hepatitis B and C in children: current treatment and future strategies. Clin Liver Dis 2006;10:133--48.
- Rogers AS, Lindsey JC, Futterman DC, et al. Serologic examination of hepatitis B infection and immunization in HIV-positive youth and associated risks. The Pediatric AIDS Clinical Trials Group Protocol 220 Team. AIDS Patient Care STDS 2000;14:651--7.
- Wang EE, King S, Goldberg E, et al. Hepatitis B and human immunodeficiency virus infection in street youths in Toronto, Canada. Pediatr Infect Dis J 1991;10:130--3.
- Mieli-Vergani G, Vergani D. Treatment of hepatitis B virus in children: why, whom, how? Indian J Gastroenterol 2006;25:121--4.
- Wasley A, Kruszon-Moran D, Kuhnert W, et al. Hepatitis B prevalence in the U.S. in the era of vaccination [abstract 723]. In: Abstracts of the Infectious Diseases Society of America 45th annual meeting; October 4--7, 2007; San Diego, CA. Arlington VA: Infectious Diseases Society of America; 2007:171.
- Toussi SS, Abadi J, Rosenberg M, et al. Prevalence of hepatitis B and C virus infections in children infected with HIV. Clin Infect Dis 2007;45:795--8.
- Tovo PA, Lazier L, Versace A. Hepatitis B virus and hepatitis C virus infections in children. Curr Opin Infect Dis 2005;18:261--6.
- Bortolotti F, Calzia R, Cadrobbi P, et al. Liver cirrhosis associated with chronic hepatitis B virus infection in childhood. J Pediatr 1986;108:224--7.
- Chen CH, Chen YY, Chen GH, et al. Hepatitis B virus transmission and hepatocarcinogenesis: a 9 year retrospective cohort of 13676 relatives with hepatocellular carcinoma. J Hepatol 2004;40:653--9.
- Bortolotti F, Guido M, Bartolacci S, et al. Chronic hepatitis B in children after e antigen seroclearance: final report of a 29-year longitudinal study. Hepatology 2006;43:556--62.
- Chen CJ, Yang HI, Su J, et al. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA 2006;295:65--73.
- Koziel MJ, Peters MG. Viral hepatitis in HIV infection. N Engl J Med 2007;356:1445--54.
- Chang MH, Hsu HY, Hsu HC, et al. The significance of spontaneous hepatitis B e antigen seroconversion in childhood: with special emphasis on the clearance of hepatitis B e antigen before 3 years of age. Hepatology 1995;22:1387--92.
- Chu CM, Hung SJ, Lin J, et al. Natural history of hepatitis B e antigen to antibody seroconversion in patients with normal serum aminotransferase levels. Am J Med 2004;116:829--34.
- Rutstein RM, Rudy B, Codispoti C, et al. Response to hepatitis B immunization by infants exposed to HIV. AIDS 1994;8:1281--4.
- Siriaksorn S, Puthanakit T, Sirisanthana T, et al. Prevalence of protective antibody against hepatitis B virus in HIV-infected children with immune recovery after highly active antiretroviral therapy. Vaccine 2006;24:3095--9.
- Shneider BL, González-Peralta R, Roberts EA. Controversies in the management of pediatric liver disease: hepatitis B, C and NAFLD. Summary of a single topic conference. Hepatology 2006;44:1344--54.
- Jain MK, Comanor L, White C, et al. Treatment of hepatitis B with lamivudine and tenofovir in HIV/HBV-coinfected patients: factors associated with response. J Viral Hepat 2007;14:176--82.
- Jonas MM. Treatment of chronic hepatitis B in children. J Pediatr Gastroenterol Nutr 2006;43(Suppl 1):S56--60.
- Heller S, Valencia-Mayoral P. Treatment of viral hepatitis in children. Arch Med Res 2007;38:702--10.
- Sokucu S, Gokçe S, Suoglu OD, et al. Comparison of interferon monotherapy with interferon-lamivudine combination treatment in children with chronic hepatitis B. Indian J Gastroenterol 2006;25:136--9.
- Kansu A, Doğanci T, Akman SA, et al. Comparison of two different regimens of combined interferon-alpha2a and lamivudine therapy in children with chronic hepatitis B infection. Antivir Ther 2006;11:255--61.
- Yilmaz A, Akcam M, Gelen T, et al. Lamivudine and high-dose interferon alpha 2a combination treatment in naïve HBeAg-positive immunoactive chronic hepatitis B in children: an East Mediterranean center's experience. Eur J Pediatr 2007;166:195--9.
- D'Antiga L, Aw M, Atkins M, et al. Combined lamivudine/interferon-alpha treatment in "immunotolerant" children perinatally infected with hepatitis B: a pilot study. J Pediatr 2006;148:228--33.
- Saltik-Temizel IN, Koçak N, Demir H. Lamivudine and high-dose interferon-alpha combination therapy for naive children with chronic hepatitis B infection. J Clin Gastroenterol 2005;39:68--70.
- Ozgenc F, Dikici B, Targan S, et al. Comparison of antiviral effect of lamivudine with interferon-alpha2a versus -alpha2b in children with chronic hepatitis B infection. Antivir Ther 2004;9:23--6.
- Marcellin P, Lau GK, Bonino F, et al. Peginterferon alfa-2a alone, lamivudine alone, and the two in combination in patients with HBeAg-negative chronic hepatitis B. N Engl J Med 2004;351:1206--17.
- Dikici B, Bosnak M, Kara IH, et al. Lamivudine and interferon-alpha combination treatment of childhood patients with chronic hepatitis B infection. Pediatr Infect Dis J 2001;20:988--92.
- Dikici G, Ozgenc F, Kalayci AG, et al. Current therapeutic approaches in childhood chronic hepatitis B infection: a multicenter study. J Gastroenterol Hepatol 2004;19:127--33.
- Kuloglu Z, Krsacloglu CT, Kansu A, et al. Liver histology of children with chronic hepatitis treated with interferon-a alone or in combination with lamivudine. J Pediatr Gastroenterol Nutr 2007;45:564--8.
- Torre D, Tambini R. Interferon-alpha therapy for chronic hepatitis B in children: a meta-analysis. Clin Infect Dis 1996;23:131--7.
- Gurakan F, Koçak N, Ozen H, et al. Comparison of standard and high dosage recombinant interferon alpha 2b for treatment of children with chronic hepatitis B infection. Pediatr Infect Dis J 2000;19:52--6.
- Yuce A, Koçak N, Ozen H, et al. Prolonged interferon alpha treatment in children with chronic hepatitis B. Ann Trop Paediatr 2001;21:77--80.
- Choe BH, Lee JH, Jang YC, et al. Long-term therapeutic efficacy of lamivudine compared with interferon-alpha in children with chronic hepatitis B: the younger the better. J Pediatr Gastroenterol Nutr 2007;44:92--8.
- Lau GK, Piratvisuth T, Luo KX, et al. Peginterferon Alfa-2a, lamivudine, and the combination for HBeAg-positive chronic hepatitis B. N Engl J Med 2005;352:2682--95.
- Schwarz KB, Mohan P, Narkewicz MR, et al. Safety, efficacy and pharmacokinetics of peginterferon alpha2a (40 kd) in children with chronic hepatitis C. J Pediatr Gastroenterol Nutr 2006;43:499--505.
- Wirth S, Pieper-Boustani H, Lang T, et al. Peginterferon alfa-2b plus ribavirin treatment in children and adolescents with chronic hepatitis C. Hepatology 2005;415.
- Baker RD, Dee D, Baker SS. Response to pegylated interferon alpha-2b and ribavirin in children with chronic hepatitis C. J Clin Gastroenterol 2007;41:111--4.
- Jonas MM, Mizerski J, Badia IB, et al. Clinical trial of lamivudine in children with chronic hepatitis B. N Engl J Med 2002;346:1706--13.
- Sokal EM, Kelly DA, Mizerski J, et al. Long-term lamivudine therapy for children with HBeAg-positive chronic hepatitis B. Hepatology 2006;43:225--32.
- Benhamou Y, Thibault V, Vig P, et al. Safety and efficacy of adefovir dipivoxil in patients infected with lamivudine-resistant hepatitis B and HIV-1. J Hepatol 2006;44:62--7.
- Locarnini S. Molecular virology and the development of resistant mutants: implications for therapy. Semin Liver Dis 2005;25(Suppl 1):9--19.
- Peters MG, Andersen J, Lynch P, et al. Randomized controlled study of tenofovir and adefovir in chronic hepatitis B virus and HIV infection: ACTG A5127. Hepatology 2006;44:1110--6.
- Lai CL, Shouval D, Lok AS, et al. Entecavir versus lamivudine for patients with HBeAg-negative chronic hepatitis B. N Engl J Med 2006;354:1011--20.
- McMahon MA, Jilek BL, Brennan TP, et al. The HBV drug entecavir---effects on HIV-1 replication and resistance. N Engl J Med 2007;356:2614--21.
- Sokal EM, Conjeevaram HS, Roberts EA, et al. Interferon alfa therapy for chronic hepatitis B in children: a multinational randomized controlled trial. Gastroenterology 1998;114:988--95.
- Gonzalez-Peralta R, Kelly DA, Haber B, et al. Interferon alfa-2b in combination with ribavirin for the treatment of chronic hepatitis C in children: efficacy, safety, and pharmacokinetics. Hepatology 2005;42:1010--8.
- Kuloglu Z, Kansu A, Berberoglu M, et al. The incidence and evolution of thyroid dysfunction during interferon-alpha therapy in children with chronic hepatitis B infection. J Pediatr Endocrinol Metab 2007;20:237--45.
- Comanor L, Minor J, Conjeevaram HS, et al. Impact of chronic hepatitis B and interferon-alpha therapy on growth of children. J Viral Hepat 2001;8:139--47.
- Jara P, Bortolotti F. Interferon-alpha treatment of chronic hepatitis B in childhood: a consensus advice based on experience in European children. J Pediatr Gastroenterol Nutr 1999;29:163--70.
- Vajro P, Migliaro F, Fontanella A, et al. Interferon: a meta-analysis of published studies in pediatric chronic hepatitis B. Acta Gastroenterol Belg 1998;61:219--23.
- Ozen H, Koçak N, Yüce A, et al. Retreatment with higher dose interferon alpha in children with chronic hepatitis B infection. Pediatr Infect Dis J 1999;18:694--7.
- Kocak N, Saltik IN, Ozen H, et al. Lamivudine treatment for children with interferon refractory chronic hepatitis B. Hepatology 2000;31:545.
- Sokal EM, Roberts EA, Mieli-Vergani G, et al. A dose ranging study of the pharmacokinetics, safety, and preliminary efficacy of lamivudine in children and adolescents with chronic hepatitis B. Antimicrob Agents Chemother 2000;44:590--7.
- Hartman C, Berkowitz D, Eshach-Adiv O, et al. Long-term lamivudine therapy for chronic hepatitis B infection in children unresponsive to interferon. J Pediatr Gastroenterol Nutr 2006;43:494--8.
- Alter MJ, Kruszon-Moran D, Nainan OV, et al. The prevalence of hepatitis C virus infection in the United States, 1988 through 1994. N Engl J Med 1999;341:556--62.
- El-Kamary SS, Serwint JR, Joffe A, et al. Prevalence of hepatitis C virus infection in urban children. J Pediatr 2003;143:54--9.
- Schuval S, Van Dyke RB, Lindsey JC, et al. Hepatitis C prevalence in children with perinatal human immunodeficiency virus infection enrolled in a long-term follow-up protocol. Arch Pediatr Adolesc Med 2004;158:1007--13.
- England K, Thorne C, Newell ML. Vertically acquired paediatric coinfection with HIV and hepatitis C virus. Lancet Infect Dis 2006;6:83--90.
- Tajiri H, Miyoshi Y, Funada S, et al. Prospective study of mother-to-infant transmission of hepatitis C virus. Pediatr Infect Dis J 2001;20:10--4.
- Jara P, Resti M, Hierro L, et al. Chronic hepatitis C virus infection in childhood: clinical patterns and evolution in 224 white children. Clin Infect Dis 2003;36:275--80.
- Murray KF, Richardson LP, Morishima C, et al. Prevalence of hepatitis C virus infection and risk factors in an incarcerated juvenile population: a pilot study. Pediatrics 2003;111:153--7.
- Cagle HH, Jacob J, Homan CE, et al. Results of a general hepatitis C lookback program for persons who received blood transfusions in a neonatal intensive care unit between January 1975 and July 1992. Arch Pediatr Adolesc Med 2007;161:125--30.
- Stramer SL. Current risks of transfusion-transmitted agents: a review. Arch Pathol Lab Med 2007;131:702--7.
- Tovo PA, Palomba E, Ferraris G, et al. Increased risk of maternal-infant hepatitis C virus transmission for women coinfected with human immunodeficiency virus type 1. Italian Study Group for HCV Infection in Children. Clin Infect Dis 1997;25:1121--4.
- Zanetti AR, Tanzi E, Romanò L, et al. A prospective study on mother-to-infant transmission of hepatitis C virus. Intervirology 1998;41:208--12.
- Gibb DM, Goodall RL, Dunn DT, et al. Mother-to-child transmission of hepatitis C virus: evidence for preventable peripartum transmission. Lancet 2000;356:904--7.
- European Paediatric Hepaptitis C Virus Network. Effects of mode of delivery and infant feeding on the risk of mother-to-child transmission of hepatitis C virus. European Paediatric Hepatitis C Virus Network. Bjog 2001;108:371--7.
- Granovsky MO, Minkoff HL, Tess BH, et al. Hepatitis C virus infection in the mothers and infants cohort study. Pediatrics 1998;102(2 Pt 1):355--9.
- Resti M, Azzari C, Mannelli F, et al. Mother to child transmission of hepatitis C virus: prospective study of risk factors and timing of infection in children born to women seronegative for HIV-1. Tuscany Study Group on Hepatitis C Virus Infection. BMJ 1998;317:437--41.
- Thomas SL, Newell ML, Peckham CS, et al. A review of hepatitis C virus (HCV) vertical transmission: risks of transmission to infants born to mothers with and without HCV viraemia or human immunodeficiency virus infection. Int J Epidemiol 1998;27:108--17.
- Mazza C, Ravaggi A, Rodella A, et al. Prospective study of mother-to-infant transmission of hepatitis C virus (HCV) infection. Study Group for Vertical Transmission. J Med Virol 1998;54:12--9.
- Okamoto M, Nagata I, Murakami J, et al. Prospective reevaluation of risk factors in mother-to-child transmission of hepatitis C virus: high virus load, vaginal delivery, and negative anti-NS4 antibody. J Infect Dis 2000;182:1511--4.
- Dal Molin G, D'Agaro P, Ansaldi F, et al. Mother-to-infant transmission of hepatitis C virus: rate of infection and assessment of viral load and IgM anti-HCV as risk factors. J Med Virol 2002;67:137--42.
- Mast EE, Hwang LY, Seto DS, et al. Risk factors for perinatal transmission of hepatitis C virus (HCV) and the natural history of HCV infection acquired in infancy. J Infect Dis 2005;192:1880--9.
- Ruiz-Extremera A, Salmerón J, Torres C, et al. Follow-up of transmission of hepatitis C to babies of human immunodeficiency virus-negative women: the role of breast-feeding in transmission. Pediatr Infect Dis J 2000;19:511--6.
- Polis CB, Shah SN, Johnson KE, et al. Impact of maternal HIV coinfection on the vertical transmission of hepatitis C virus: a meta-analysis. Clin Infect Dis 2007;44:1123--31.
- Mok J, Pembrey L, Tovo PA, et al. When does mother to child transmission of hepatitis C virus occur? Arch Dis Child Fetal Neonatal Ed 2005;90:F156--60.
- Conte D, Fraquelli M, Prati D, et al. Prevalence and clinical course of chronic hepatitis C virus (HCV) infection and rate of HCV vertical transmission in a cohort of 15,250 pregnant women. Hepatology 2000;31:751--5.
- Papaevangelou V, Pollack H, Rochford G, et al. Increased transmission of vertical hepatitis C virus (HCV) infection to human immunodeficiency virus (HIV)-infected infants of HIV- and HCV-coinfected women. J Infect Dis 1998;178:1047--52.
- Marine-Barjoan E, Berrébi A, Giordanengo V, et al. HCV/HIV co-infection, HCV viral load and mode of delivery: risk factors for mother-to-child transmission of hepatitis C virus? AIDS 2007;21:1811--5.
- European Paediatric Hepatitis C Virus Network. A significant sex---but not elective cesarean section---effect on mother-to-child transmission of hepatitis C virus infection. J Infect Dis 2005;192:1872--9.
- Lin HH, Kao JH, Hsu HY, et al. Absence of infection in breast-fed infants born to hepatitis C virus-infected mothers. J Pediatr 1995;126:589--91.
- Pappalardo B. Influence of maternal human immunodeficiency virus (HIV) co-infection on vertical transmission of hepatitis C virus (HCV): a meta-analysis. Int J Epidemiol 2003;32:727--34.
- Thomas DL, Villano SA, Riester KA, et al. Perinatal transmission of hepatitis C virus from human immunodeficiency virus type 1-infected mothers. Women and Infants Transmission Study. J Infect Dis 1998;177:1480--8.
- Paccagini S, Principi N, Massironi E, et al. Perinatal transmission and manifestation of hepatitis C virus infection in a high risk population. Pediatr Infect Dis J 1995;14:195--9.
- Hershow RC, Riester KA, Lew J, et al. Increased vertical transmission of human immunodeficiency virus from hepatitis C virus-coinfected mothers. Women and Infants Transmission Study. J Infect Dis 1997;176:414--20.
- Nigro G, D'Orio F, Catania S, et al. Mother to infant transmission of coinfection by human immunodeficiency virus and hepatitis C virus: prevalence and clinical manifestations. Arch Virol 1997;142:453--7.
- Giovannini M, Tagger A, Ribero ML, et al. Maternal-infant transmission of hepatitis C virus and HIV infections: a possible interaction. Lancet 1990;335:1166.
- Scott JD, Gretch DR. Molecular diagnostics of hepatitis C virus infection: a systematic review. JAMA 2007;297:724--32.
- El Sherbini A, Hassan W, Abdel-Hamid M, et al. Natural history of hepatitis C virus among apparently normal schoolchildren: follow-up after 7 years. J Trop Pediatr 2003;49:384--5.
- Rerksuppaphol S, Hardikar W, Dore GJ. Long-term outcome of vertically acquired and post-transfusion hepatitis C infection in children. J Gastroenterol Hepatol 2004;19:1357--62.
- England K, Pembrey L, Tovo PA, et al. Growth in the first 5 years of life is unaffected in children with perinatally acquired hepatitis C infection. J Pediatr 2005;147:227--32.
- Resti M, Jara P, Hierro L, et al. Clinical features and progression of perinatally acquired hepatitis C virus infection. J Med Virol 2003;70:373--7.
- European Paediatric Hepatitis C Virus Network. Three broad modalities in the natural history of vertically acquired hepatitis C virus infection. Clin Infect Dis 2005;41:45--51.
- Bortolotti F, Resti M, Marcellini M, et al. Hepatitis C virus (HCV) genotypes in 373 Italian children with HCV infection: changing distribution and correlation with clinical features and outcome. Gut 2005;54:852--7.
- Guido M, Rugge M, Jara P, et al. Chronic hepatitis C in children: the pathological and clinical spectrum. Gastroenterology 1998;115:1525--9.
- Aach RD, Yomtovian RA, Hack M. Neonatal and pediatric posttransfusion hepatitis C: a look back and a look forward. Pediatrics 2000;105(4 Pt 1):836--42.
- Mohan P, Colvin C, Glymph C, et al. Clinical spectrum and histopathologic features of chronic hepatitis C infection in children. J Pediatr 2007;150:168--74, 174.e1.
- Davison SM, Mieli-Vergani G, Sira J, et al. Perinatal hepatitis C virus infection: diagnosis and management. Arch Dis Child 2006;91:781--5.
- Tovo PA, Pembrey LJ, Newell ML. Persistence rate and progression of vertically acquired hepatitis C infection. European Paediatric Hepatitis C Virus Infection. J Infect Dis 2000;181:419--24.
- Graham CS, Baden LR, Yu E, et al. Influence of human immunodeficiency virus infection on the course of hepatitis C virus infection: a meta-analysis. Clin Infect Dis 2001;33:562--9.
- Merchante N, Girón-González JA, González-Serrano M, et al. Survival and prognostic factors of HIV-infected patients with HCV-related end-stage liver disease. AIDS 2006;20:49--57.
- Shivraj SO, Chattopadhya D, Grover G, et al. Role of HCV coinfection towards disease progression and survival in HIV-1 infected children: a follow-up study of 10 years. J Trop Pediatr 2006;52:206--11.
- Dunn DT, Gibb DM, Healy M, et al. Timing and interpretation of tests for diagnosing perinatally acquired hepatitis C virus infection. Pediatr Infect Dis J 2001;20:715--6.
- Polywka S, Pembrey L, Tovo PA, et al. Accuracy of HCV-RNA PCR tests for diagnosis or exclusion of vertically acquired HCV infection. J Med Virol 2006;78:305--10.
- Perinatal HIV Guidelines Working Group. Public Health Service Task Force Recommendations for use of antiretroviral drugs in pregnant HIV-infected women for mternal health and interventions to reduce perinatal HIV-1 transmission in the United States. July 8, 2008:1--98. Available at http://aidsinfo.nih.gov/ContentFiles/PerinatalGL.pdf.
- CDC. Recommendations for prevention and control of hepatitis C virus (HCV) infection and HCV-related chronic disease. MMWR 1998;47(No. RR-19).
- Strader DB, Wright T, Thomas DL, et al. Diagnosis, management, and treatment of hepatitis C. Hepatology 2004;39:1147--71.
- Soriano V, Puoti M, Sulkowski M, et al. Care of patients coinfected with HIV and hepatitis C virus: 2007 updated recommendations from the HCV-HIV International Panel. AIDS 2007;21:1073--89.
- Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med 2002;347:975--82.
- Chung RT, Andersen J, Volberding P, et al. Peginterferon alfa-2a plus ribavirin versus interferon alfa-2a plus ribavirin for chronic hepatitis C in HIV-coinfected persons. N Engl J Med 2004;351:451--9.
- Narkewicz MR, Cabrera R, Gonzalez-Peralta RP. The "C" of viral hepatitis in children. Semin Liver Dis 2007;27:295--311.
- Christensson B, Wiebe T, Akesson A, et al. Interferon-alpha and ribavirin treatment of hepatitis C in children with malignancy in remission. Clin Infect Dis 2000;30:585--6.
- Lackner H, Moser A, Deutsch J, et al. Interferon-alpha and ribavirin in treating children and young adults with chronic hepatitis C after malignancy. Pediatrics 2000;106:E53.
- Kowala-Piaskowska A, Sluzewski W, Figlerowicz M, et al. Early virological response in children with chronic hepatitis C treated with pegylated interferon and ribavirin. Infection 2007;35:175--9.
- Wirth S, Lang T, Gehring S, et al. Recombinant alfa-interferon plus ribavirin therapy in children and adolescents with chronic hepatitis C. Hepatology 2002;36:1280--4.
- Caserta MT, McDermott MP, Dewhurst S, et al. Human herpesvirus 6 (HHV6) DNA persistence and reactivation in healthy children. J Pediatr 2004;145:478--84.
- Kositanont U, Wasi C, Wanprapar N, et al. Primary infection of human herpesvirus 6 in children with vertical infection of human immunodeficiency virus type 1. J Infect Dis 1999;180:50--5.
- Zerr D, Meier AS, Selke SS, et al. A population-based study of primary human herpesvirus 6 infection. N Engl J Med 2005;352:768--76.
- Hall CB, Caserta MT, Schnabel KC, et al. Congenital infections with human herpesvirus 6 (HHV6) and human herpesvirus 7 (HHV7). J Pediatr 2004;145:472--7.
- Pruksananonda P, Hall CB, Insel RA, et al. Primary human herpesvirus 6 infection in young children. N Engl J Med 1992;326:1445--50.
- Okuno T, Oishi H, Hayashi K, et al. Human herpesviruses 6 and 7 in cervixes of pregnant women. J Clin Microbiol 1995;33:1968--70.
- Bailllargeon J, Piper J, Leach CT. Epidemiology of human herpesvirus 6 (HHV-6) infection in pregnant and nonpregnant women. J Clin Virol 2000;16:149--57.
- Adams O, Krempe C, Kögler G, et al. Congenital infections with human herpesvirus 6. J Infect Dis 1998;178:544--6.
- Dahl H, Fjaertoft G, Norsted T, et al. Reactivation of human herpesvirus 6 during pregnancy. J Infect Dis 1999;180:2035--8.
- Horvat RT, Wood C, Josephs SF, et al. Transactivation of the human immunodeficiency virus promoter by human herpesvirus 6 (HHV-6) strains GS and Z-29 in primary human T lymphocytes and identification of transactivating HHV-6(GS) gene fragments. J Virol 1991;65:2895--902.
- Isegawa Y, Katahira J, Yamanishi K, et al. Reactivation of latent human immunodeficiency virus 1 by human herpesvirus 6 infection. Acta Virol 2007;51:13--20.
- Fujiwara N, Namba H, Ohuchi R, et al. Monitoring of human herpesvirus-6 and -7 genomes in saliva samples of healthy adults by competitive quantitative PCR. J Med Virol 2000;61:208--13.
- Hall CB, Long CE, Schnabel KC, et al. Human herpesvirus-6 infection in children. A prospective study of complications and reactivation. N Engl J Med 1994;331:342--8.
- Asano Y, Yoshikawa T, Suga S, et al. Clinical features of infants with primary human herpesvirus 6 infection (exanthem subitum, roseola infantum). Pediatrics 1994;93:104--8.
- Suga S, Yoshikawa T, Asano Y, et al. Clinical and virological analyses of 21 infants with exanthem subitum (roseola infantum) and central nervous system complications. Ann Neurol 1993;33:597--603.
- Isaacson E, Glaser CA, Forghani B, et al. Evidence of human herpesvirus 6 infection in 4 immunocompetent patients with encephalitis. Clin Infect Dis 2005;40:890--3.
- Mannonen L, Herrgård E, Valmari P, et al. Primary human herpesvirus-6 infection in the central nervous system can cause severe disease. Pediatr Neurol 2007;37:186--91.
- Caserta MT, Mock DJ, Dewhurst S. Human herpesvirus 6. Clin Infect Dis 2001;33:829--33.
- Zerr DM, Frenkel LM, Huang ML, et al. Polymerase chain reaction diagnosis of primary human herpesvirus-6 infection in the acute care setting. J Pediatr 2006;149:480--5.
- Leach CT. Human herpesvirus-6 and -7 infections in children: agents of roseola and other syndromes. Curr Opin Pediatr 2000;12:269--74.
- Rappaport D, Engelhard D, Tagger G, et al. Antiviral prophylaxis may prevent human herpesvirus-6 reactivation in bone marrow transplant recipients. Transpl Infect Dis 2002;4:10--6.
- Tokimassa S, Hara J, Osugi Y, et al. Ganciclovir is effective for prophylaxis and treatment of human herpesvirus-6 in allogeneic stem cell transplantation. Bone Marrow Transplant 2002;29:595--8.
- Burns WH, Sanford GR. Susceptibility of human herpesvirus 6 to antivirals in vitro. J Infect Dis 1990;162:634--7.
- De Clercq E, Naesens L, De Bolle L, et al. Antiviral agents active against human herpesviruses HHV-6, HHV-7 and HHV-8. Rev Med Virol 2001;11:381--95.
- Rieux C, Gautheret-Dejean A, Challine-Lehmann D, et al. Human herpesvirus-6 meningoencephalitis in a recipient of an unrelated allogeneic bone marrow transplantation. Transplantation 1998;65:1408--11.
- Wang FZ, Linde A, Hägglund H, et al. Human herpesvirus 6 DNA in cerebrospinal fluid specimens from allogeneic bone marrow transplant patients: does it have clinical significance? Clin Infect Dis 1999;28:562--8.
- Singh N, Paterson DL. Encephalitis caused by human herpesvirus-6 in transplant recipients: relevance of a novel neurotropic virus. Transplantation 2000;69:2474--9.
- Drobyski WR, Knox KK, Majewski D, et al. Brief report: fatal encephalitis due to variant B human herpesvirus-6 infection in a bone marrow-transplant recipient. N Engl J Med 1994;330:1356--60.
- Pöhlmann C, Schetelig J, Reuner U, et al. Cidofovir and foscarnet for treatment of human herpesvirus 6 encephalitis in a neutropenic stem cell transplant recipient. Clin Infect Dis 2007;44:e118--20.
- Janoly-Dumenil A, Galambrun C, Basset T, et al. Human herpes virus-6 encephalitis in a paediatric bone marrow recipient: successful treatment with pharmacokinetic monitoring and high doses of ganciclovir. Bone Marrow Transplant 2006;38:769--70.
- De Bolle L, Manichanh C, Agut H, et al. Human herpesvirus 6 DNA polymerase: enzymatic parameters, sensitivity to ganciclovir and determination of the role of the A961V mutation in HHV-6 ganciclovir resistance. Antiviral Res 2004;64:17--25.
- Gao SJ, Kingsley L, Hoover DR, et al. Seroconversion to antibodies against Kaposi's sarcoma-associated herpesvirus-related latent nuclear antigens before the development of Kaposi's sarcoma. N Engl J Med 1996;335:233--41.
- Lennette ET, Blackbourn DJ, Levy JA. Antibodies to human herpesvirus type 8 in the general population and in Kaposi's sarcoma patients. Lancet 1996;348:858--61.
- Engels EA, Atkinson JO, Graubard BI, et al. Risk factors for human herpesvirus 8 infection among adults in the United States and evidence for sexual transmission. J Infect Dis 2007;196:199--207.
- Whitby D, Smith NA, Matthews S, et al. Human herpesvirus 8: seroepidemiology among women and detection in the genital tract of seropositive women. J Infect Dis 1999;179:234--6.
- Goedert JJ, Kedes DH, Ganem D. Antibodies to human herpesvirus 8 in women and infants born in Haiti and the USA. Lancet 1997;349:1368.
- Huang LM, Huang SY, Chen MY, et al. Geographical differences in human herpesvirus 8 seroepidemiology: a survey of 1,201 individuals in Asia. J Med Virol 2000;60:290--3.
- Serraino D, Locatelli M, Songini M, et al. Human herpes virus-8 infection among pregnant women and their children: results from the Sardinia-IDDM Study 2. Int J Cancer 2001;91:740--1.
- Casper C, Krantz E, Selke S, et al. Frequent and asymptomatic oropharyngeal shedding of human herpesvirus 8 among immunocompetent men. J Infect Dis 2007;195:30--6.
- Mbulaiteye SM, Pfeiffer RM, Whitby D, et al. Human herpesvirus 8 infection within families in rural Tanzania. J Infect Dis 2003;187:1780--5.
- He J, Bhat G, Kankasa C, et al. Seroprevalence of human herpesvirus 8 among Zambian women of childbearing age without Kaposi's sarcoma (KS) and mother-child pairs with KS. J Infect Dis 1998;178:1787--90.
- Gessain A, Mauclere P, van Beveren M, et al. Human herpesvirus 8 primary infection occurs during childhood in Cameroon, Central Africa. Int J Cancer 1999;81:189--92.
- Sitas F, Newton R, Boshoff C. Increasing probability of mother-to-child transmission of HHV-8 with increasing maternal antibody titer for HHV-8. N Engl J Med 1999;340:1923.
- Calabro ML, Gasperini P, Barbierato M, et al. A search for human herpesvirus 8 (HHV-8) in HIV-1 infected mothers and their infants does not suggest vertical transmission of HHV-8. Int J Cancer 2000;85:296--7.
- Plancoulaine S, Abel L, van Beveren M, et al. Human herpesvirus 8 transmission from mother to child and between siblings in an endemic population. Lancet 2000;356:1062--5.
- Malope BI, Pfeiffer RM, Mbisa G, et al. Transmission of Kaposi sarcoma-associated herpesvirus between mothers and children in a South African population. J Acquir Immune Defic Syndr 2007;44:351--5.
- Casper C, Meier AS, Wald A, et al. Human herpesvirus 8 infection among adolescents in the REACH cohort. Arch Pediatr Adolesc Med 2006;160:937--42.
- Cannon MJ, Dollard SC, Smith DK, et al. Blood-borne and sexual transmission of human herpesvirus 8 in women with or at risk for human immunodeficiency virus infection. N Engl J Med 2001;344:637--43.
- Hladik W, Dollard SC, Mermin J, et al. Transmission of human herpesvirus 8 by blood transfusion. N Engl J Med 2006;355:1331--8.
- Lisco A, Barbierato M, Fiore JR, et al. Pregnancy and human herpesvirus 8 reactivation in human immunodeficiency virus type 1-infected women. J Clin Microbiol 2006;44:3863--71.
- Glesby MJ, Hoover DR, Weng S, et al. Use of antiherpes drugs and the risk of Kaposi's sarcoma: data from the Multicenter AIDS Cohort Study. J Infect Dis 1996;173:1477--80.
- Mocroft A, Youle M, Gazzard B, et al. Anti-herpesvirus treatment and risk of Kaposi's sarcoma in HIV infection. Royal Free/Chelsea and Westminster Hospitals Collaborative Group. AIDS 1996;10:1101--5.
- Cannon JS, Hamzeh F, Moore S, et al. Human herpesvirus 8-encoded thymidine kinase and phosphotransferase homologues confer sensitivity to ganciclovir. J Virol 1999;73:4786--93.
- Neyts J, De Clercq E. Antiviral drug susceptibility of human herpesvirus 8. Antimicrob Agents Chemother 1997;41:2754--6.
- Kedes DH, Ganem D. Sensitivity of Kaposi's sarcoma-associated herpesvirus replication to antiviral drugs. Implications for potential therapy. J Clin Invest 1997;99:2082--6.
- Robles R, Lugo D, Gee L, et al. Effect of antiviral drugs used to treat cytomegalovirus end-organ disease on subsequent course of previously diagnosed Kaposi's sarcoma in patients with AIDS. J Acquir Immune Defic Syndr Hum Retrovirol 1999;20:34--8.
- Cannon MJ, Laney AS, Pellett PE. Human herpesvirus 8: current issues. Clin Infect Dis 2003;37:82--7.
- Ledergerber B, Egger M, Erard V, et al. AIDS-related opportunistic illnesses occurring after initiation of potent antiretroviral therapy: the Swiss HIV Cohort Study. JAMA 1999;282:2220--6.
- Ziegler JL, Katongole-Mbidde E. Kaposi's sarcoma in childhood: an analysis of 100 cases from Uganda and relationship to HIV infection. Int J Cancer 1996;65:200--3.
- Andreoni M, Sarmati L, Nicastri E, et al. Primary human herpesvirus 8 infection in immunocompetent children. JAMA 2002;287:1295--300.
- Luppi M, P. B, TF. S, et al. Bone marrow failure associated with human herpesvirus 8 infection after transplantation. N Engl J Med 2000;343:1378--85.
- Luppi M, Barozzi P, Rasini V, et al. Severe pancytopenia and hemophagocytosis after HHV-8 primary infection in a renal transplant patient successfully treated with foscarnet. Transplantation 2002;74:131--2.
- Bhaduri-McIntosh S. Human herpesvirus-8: clinical features of an emerging viral pathogen. Pediatr Infect Dis J 2005;24:81--2.
- Laney AS, Cannon MJ, Jaffe HW, et al. Human herpesvirus 8 presence and viral load are associated with the progression of AIDS-associated Kaposi's sarcoma. AIDS 2007;21:1541--5.
- Anderson LA, Goedert JJ. Tumor markers and treatments for Kaposi sarcoma. AIDS 2007;21:1637--9.
- Klass CM, Offermann MK. Targeting human herpesvirus-8 for treatment of Kaposi's sarcoma and primary effusion lymphoma. Curr Opin Oncol 2005;17:447--55.
- Casper C, Nichols WG, Huang ML, et al. Remission of HHV-8 and HIV-associated multicentric Castleman disease with ganciclovir treatment. Blood 2004;103:1632--4.
- Aboulafia DM. Interleukin-2, ganciclovir, and high-dose zidovudine for the treatment of AIDS-associated primary central nervous system lymphoma. Clin Infect Dis 2002;34:1660--2.
- Crum-Cianflone NF, Wallace MR, Looney D. Successful secondary prophylaxis for primary effusion lymphoma with human herpesvirus 8 therapy. AIDS 2006;20:1567--9.
- Leidner RS, Aboulafia DM. Recrudescent Kaposi's sarcoma after initiation of HAART: a manifestation of immune reconstitution syndrome. AIDS Patient Care STDS 2005;19:635--44.
- Bower M, Nelson M, Young AM, et al. Immune reconstitution inflammatory syndrome associated with Kaposi's sarcoma. J clin Oncol 2005;23:5224--8.
- Whitley R, Kimberlin DW, Roizman B. Herpes simplex viruses. Clin Infect Dis 1998;26:541--53.
- Xu F, Lee FK, Morrow RA, et al. Seroprevalence of herpes simplex virus type 1 in children in the United States. J Pediatr 2007;151:374--7.
- Xu F, Sternberg MR, Kottiri BJ, et al. Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. JAMA 2006;296:964--73.
- Fleming DT, McQuillan GM, Johnson RE, et al. Herpes simplex virus type 2 in the United States, 1976 to 1994. N Engl J Med 1997;337:1105--11.
- Whitley R, Davis EA, Suppapanya N. Incidence of neonatal herpes simplex virus infections in a managed-care population. Sex Transm Dis 2007;34:704--8.
- Prober CG, Corey L, Brown ZA, et al. The management of pregnancies complicated by genital infections with herpes simplex virus. Clin Infect Dis 1992;15:1031--8.
- Brown ZA, Wald A, Morrow RA, et al. Effect of serologic status and cesarean delivery on transmission rates of herpes simplex virus from mother to infant. JAMA 2003;289:203--9.
- Xu F, Markowitz LE, Gottlieb SL, et al. Seroprevalence of herpes simplex virus types 1 and 2 in pregnant women in the United States. Am J Obstet Gynecol 2007;196:43.e1--6.
- Augenbraun M, Feldman J, Chirgwin K, et al. Increased genital shedding of herpes simplex virus type 2 in HIV-seropositive women. Ann Intern Med 1995;123:845--7.
- Catalano PM, Merritt AO, Mead PB. Incidence of genital herpes simplex virus at the time of delivery in women with known risk factors. Am J Obstet Gynecol 1991;164:1303--6.
- Hitti J, Watts DH, Burchett SK, et al. Herpes simplex virus seropositivity and reactivation at delivery among pregnant women infected with human immunodeficiency virus-1. Am J Obstet Gynecol 1997;177:450--4.
- Drake AL, John-Stewart GC, Wald A, et al. Herpes simplex virus type 2 and risk of intrapartum human immunodeficiency virus transmission. Obstet Gynecol 2007;109(2 Pt 1):403--9.
- Salvini F, Carminati G, Pinzani R, et al. Chronic ulcerative herpes simplex virus infection in HIV-infected children. AIDS Patient Care STDS 1997;11:421--8.
- Kimberlin DW, Lin CY, Jacobs RF, et al. Natural history of neonatal herpes simplex virus infections in the acyclovir era. Pediatrics 2001;108:223--9.
- Kimberlin DW. Herpes simplex virus infections of the newborn. Semin Perinatol 2007;31:19--25.
- Gutierrez K, Arvin AM. Long term antiviral suppression after treatment for neonatal herpes infection. Pediatr Infect Dis J 2003;22:371--2.
- Kimberlin DW, Powell D, Gruber W, et al. Administration of oral acyclovir suppressive therapy after neonatal herpes simplex virus disease limited to the skin, eyes and mouth: results of a phase I/II trial. Pediatr Infect Dis J 1996;15:247--54.
- Corey L, Adams HG, Brown ZA, et al. Genital herpes simplex virus infections: clinical manifestations, course, and complications. Ann Intern Med 1983;98:958--72.
- Kimberlin DW, Lakeman FD, Arvin AM, et al. Application of the polymerase chain reaction to the diagnosis and management of neonatal herpes simplex virus disease. National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. J Infect Dis 1996;174:1162--7.
- Kimura H, Aso K, Kuzushima K, et al. Relapse of herpes simplex encephalitis in children. Pediatrics 1992;89(5 Pt 1):891--4.
- Posavad CM, Wald A, Kuntz S, et al. Frequent reactivation of herpes simplex virus among HIV-1-infected patients treated with highly active antiretroviral therapy. J Infect Dis 2004;190:693--6.
- Scott LL, Hollier LM, McIntire D, et al. Acyclovir suppression to prevent recurrent genital herpes at delivery. Infect Dis Obstet Gynecol 2002;10:71--7.
- Sheffield JS, Hollier LM, Hill JB, et al. Acyclovir prophylaxis to prevent herpes simplex virus recurrence at delivery: a systematic review. Obstet Gynecol 2003;102:1396--403.
- Sheffield JS, Hill JB, Hollier LM, et al. Valacyclovir prophylaxis to prevent recurrent herpes at delivery: a randomized clinical trial. Obstet Gynecol 2006;108:141--7.
- Kimberlin DW. Herpes simplex virus infections in neonates and early childhood. Semin Pediatr Infect Dis 2005;16:271--81.
- Haddad J, Langer B, Astruc D, et al. Oral acyclovir and recurrent genital herpes during late pregnancy. Obstet Gynecol 1993;82:102--4.
- American College of Obstetricians and Gynecologists. Management of herpes in pregnancy. ACLG Practice Bulletin 8. Washington DC: American College of Obstetricians and Gynecologists; 1999.
- Strick LB, Wald A, Celum C. Management of herpes simplex virus type 2 infection in HIV type 1-infected persons. Clin Infect Dis 2006;43:347--56.
- Kimberlin DW, Lin CY, Jacobs RF, et al. Safety and efficacy of high-dose intravenous acyclovir in the management of neonatal herpes simplex virus infections. Pediatrics 2001;108:230--8.
- Balfour HH, Jr. Antiviral drugs. N Engl J Med 1999;340:1255--68.
- Feinberg JE, Hurwitz S, Cooper D, et al. A randomized, double-blind trial of valaciclovir prophylaxis for cytomegalovirus disease in patients with advanced human immunodeficiency virus infection. AIDS Clinical Trials Group Protocol 204/Glaxo Wellcome 123-014 International CMV Prophylaxis Study Group. J Infect Dis 1998;177:48--56.
- Schacker T, Hu HL, Koelle DM, et al. Famciclovir for the suppression of symptomatic and asymptomatic herpes simplex virus reactivation in HIV-infected persons. A double-blind, placebo-controlled trial. Ann Intern Med 1998;128:21--8.
- Aoki FY, Tyring S, Diaz-Mitoma F, et al. Single-day, patient-initiated famciclovir therapy for recurrent genital herpes: a randomized, double-blind, placebo-controlled trial. Clin Infect Dis 2006;42:8--13.
- Spruance S, Bodsworth N, Resnick H, et al. Single-dose, patient-initiated famciclovir: a randomized, double-blind, placebo-controlled trial for episodic treatment of herpes labialis. J Am Acad Dermatol 2006;55:47--53.
- Eksborg S, Pal N, Kalin M, et al. Pharmacokinetics of acyclovir in immunocompromized children with leukopenia and mucositis after chemotherapy: can intravenous acyclovir be substituted by oral valacyclovir? Med Pediatr Oncol 2002;38;240--6.
- Eksborg S. The pharmacokinetics of antiviral therapy in paediatric patients. Herpes 2003;10:66--71.
- Balfour HH, Jr, Benson C, Braun J, et al. Management of acyclovir-resistant herpes simplex and varicella-zoster virus infections. J Acquir Immune Defic Syndr 1994;7:254--60.
- Lateef F, Don PC, Kaufmann M, et al. Treatment of acyclovir-resistant, foscarnet-unresponsive HSV infection with topical cidofovir in a child with AIDS. Arch Dermatol 1998;134:1169--70.
- Blot N, Schneider P, Young P, et al. Treatment of an acyclovir and foscarnet-resistant herpes simplex virus infection with cidofovir in a child after an unrelated bone marrow transplant. Bone Marrow Transplant 2000;26:903--5.
- Munoz N, Castellsagué X, de González AB, et al. Chapter 1: HPV in the etiology of human cancer. Vaccine 2006;24(S3):S1--S10.
- Gutman LT, St Claire K, Herman-Giddens ME, et al. Evaluation of sexually abused and nonabused young girls for intravaginal human papillomavirus infection. Am J Dis Child 1992;146:694--9.
- Beutner KR, Reitano MV, Richwald GA, et al. External genital warts: report of the American Medical Association Consensus Conference. AMA Expert Panel on External Genital Warts. Clin Infect Dis 1998;27:796--806.
- Rintala M, Grénman SE, Järvenkylä ME, et al. High-risk types of human papillomavirus (HPV) DNA in oral and genital mucosa of infants during their first 3 years of life: experience from the Finnish HPV Family Study. Clin Infect Dis 2005;41:1728--33.
- Smith EM, Ritchie JM, Yankowitz J, et al. Human papillomavirus prevalence and types in newborns and parents: concordance and modes of transmission. Sex Transm Dis 2004;31:57--62.
- Hernandez-Giron C, Smith JS, Lorincz A, et al. High-risk human papillomavirus detection and related risk factors among pregnant and nonpregnant women in Mexico. Sex Transm Dis 2005;32:613--8.
- Chan PK, Chang AR, Tam WH, et al. Prevalence and genotype distribution of cervical human papillomavirus infection: comparison between pregnant women and non-pregnant controls. J Med Virol 2002;67:583--8.
- Fife KH, Katz BP, Brizendine EJ, et al. Cervical human papillomavirus deoxyribonucleic acid persists throughout pregnancy and decreases in the postpartum period. Am J Obstet Gynecol 1999;180:1110--4.
- Hagensee ME, Cameron JE, Leigh JE, et al. Human papillomavirus infection and disease in HIV-infected individuals. Am J Med Sci 2004;328:57--63.
- Moscicki AB, Ellenberg JH, Vermund SH, et al. Prevalence of and risks for cervical human papillomavirus infection and squamous intraepithelial lesions in adolescent girls: impact of infection with human immunodeficiency virus. Arch Pediatr Adolesc Med 2000;154:127--34.
- Bollen LJ, Chuachoowong R, Kilmarx PH, et al. Human papillomavirus (HPV) detection among human immunodeficiency virus-infected pregnant Thai women: implications for future HPV immunization. Sex Transm Dis 2006;33:259--64.
- Armbruster-Moraes E, Ioshimoto LM, Leão E, et al. Presence of human papillomavirus DNA in amniotic fluids of pregnant women with cervical lesions. Gynecol Oncol 1994;54:152--8.
- Tseng CJ, Lin CY, Wang RL, et al. Possible transplacental transmission of human papillomaviruses. Am J Obstet Gynecol 1992;166(1 Pt 1):35--40.
- Tseng CJ, Liang CC, Soong YK, et al. Perinatal transmission of human papillomavirus in infants: relationship between infection rate and mode of delivery. Obstet Gynecol 1998;91:92--6.
- Tenti P, Zappatore R, Migliora P, et al. Perinatal transmission of human papillomavirus from gravidas with latent infections. Obstet Gynecol 1999;93:475--9.
- Smith EM, Swarnavel S, Ritchie JM, et al. Prevalence of human papillomavirus in the oral cavity/oropharynx in a large population of children and adolescents. Pediatr Infect Dis J 2007;26:836--40.
- Moscicki AB, Hills N, Shiboski S, et al. Risks for incident human papillomavirus infection and low-grade squamous intraepithelial lesion development in young females. JAMA 2001;285:2995--3002.
- Burchell AN, Winer RL, de Sanjosé S, et al. Epidemiology and transmission dynamics of genital HPV infection. Vaccine 2006;24:S52--61.
- Winer RL, Lee SK, Hughes JP, et al. Genital human papillomavirus infection: incidence and risk factors in a cohort of female university students. Am J Epidemiol 2003;157:218--26.
- Winer RL, Hughes JP, Feng Q, et al. Condom use and the risk of genital human papillomavirus infection in young women. N Engl J Med 2006;354:2645--54.
- Munoz N, Méndez F, Posso H, et al. Incidence, duration, and determinants of cervical human papillomavirus infection in a cohort of Colombian women with normal cytological results. J Infect Dis 2004;190:2077--87.
- Brown DR, Shew ML, Qadadri B, et al. A longitudinal study of genital human papillomavirus infection in a cohort of closely followed adolescent women. J Infect Dis 2005;191:182--92.
- Moscicki AB, Shiboski S, Broering J, et al. The natural history of human papillomavirus infection as measured by repeated DNA testing in adolescent and young women. J Pediatr 1998;132:277--84.
- Tarkowski TA, Koumans EH, Sawyer M, et al. Epidemiology of human papillomavirus infection and abnormal cytologic test results in an urban adolescent population. J Infect Dis 2004;189:46--50.
- Winer RL, Feng Q, Hughes JP, et al. Risk of female human papillomavirus acquisition associated with first male sex partner. J Infect Dis 2008;197:279--82.
- Ho GY, Bierman R, Beardsley L, et al. Natural history of cervicovaginal papillomavirus infection in young women. N Engl J Med 1998;338:423--8.
- Strickler HD, Burk RD, Fazzari M, et al. Natural history and possible reactivation of human papillomavirus in human immunodeficiency virus-positive women. J Natl Cancer Inst 2005;97:577--86.
- Moscicki AB, Ellenberg JH, Farhat S, et al. Persistence of human papillomavirus infection in HIV-infected and HIV-uninfected adolescent girls: risk factors and differences, by phylogenetic type. J Infect Dis 2004;190:37--45.
- Moscicki AB, Schiffman M, Kjaer S, et al. Updating the natural history of HPV and anogenital cancer. Vaccine 2006;24(Suppl 3):S42--51.
- Moscicki AB, Durako SJ, Houser J, et al. Human papillomavirus infection and abnormal cytology of the anus in HIV-infected and uninfected adolescents. AIDS 2003;17:311--20.
- Palefsky J. HPV infection and HPV-associated neoplasia in immunocompromised women. Int J Gynaecol Obstet 2006;94(Suppl 1):S56-64.
- Palefsky J, Holly EA, Ralston ML, et al. Prevalence and risk factors for anal human papillomavirus infection in human immunodeficiency virus (HIV)-positive and high-risk HIV-negative women. J Infect Dis 2001;183:383--91.
- Frisch M, Biggar RJ, Goedert JJ. Human papillomavirus-associated cancers in patients with human immunodeficiency virus infection and acquired immunodeficiency syndrome. J Natl Cancer Inst 2000;92:1500--10.
- Moscicki A, Burt VG, Kanowitz S, et al. The significance of squamous metaplasia in the development of low grade squamous intraepithelial lesions in young women. Cancer 1999;85:1139--44.
- Moscicki AB, Ellenberg JH, Crowley-Nowick P, et al. Risk of high-grade squamous intraepithelial lesion in HIV-infected adolescents. J Infect Dis 2004;190:1413--21.
- Arbyn M, Sasieni P, Meijer CJ, et al. Clinical applications of HPV testing: a summary of meta-analyses. Vaccine 2006;24(Suppl 3):S78--89.
- Wright TC, Jr, Massad LS, Dunton CJ, et al. 2006 consensus guidelines for the management of women with cervical intraepithelial neoplasia or adenocarcinoma in situ. Am J Obstet Gynecol 2007;197:340--5.
- The FUTURE II Study Group. Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N Engl J Med 2007;356:1915--27.
- Garland S, Hernandez-Avila M, Wheeler CM, et al. Quadrivalent vaccine against human papillomavirus to prevent anogenital diseases. N Engl J Med 2007;356:1928--43.
- Paavonen J, Jenkins D, Bosch FX, et al. Efficacy of a prophylactic adjuvanted bivalent L1 virus-like-particle vaccine against infection with human papillomavirus types 16 and 18 in young women: an interim analysis of a phase III double-blind, randomised controlled trial. Lancet 2007;369:2161--70.
- CDC. Quadrivalent human papillomavirus vaccine: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR 2007;56(No. RR-2). Available at http://www.cdc.gov/mmwr/pdf/rr/rr5602.pdf. Accessed June 13, 2009.
- Weinberg A, Song LY, Handelsman E, et al. Safety and immunogenicity of a quadrivalent vaccine to prevent human papilloma virus (HPV) infection in HIV-infected children: IMPAACT P1047. 15th Conference on Retroviruses and Opportunistic Infections, Feb 3--6, 2008, Boston MA, paper 619a.
- Goldie SJ, Kuntz KM, Weinstein MC, et al. The clinical effectiveness and cost-effectiveness of screening for anal squamous intraepithelial lesions in homosexual and bisexual HIV-positive men. JAMA 1999;281:1822--9.
- Konstantinopoulos P, Schlecht HP, Bryan B, et al. HIV-associated anal squamous cell cancer: an otherwise preventable disease. J Clin Oncol 2006;24:4516--7.
- De Panfilis G, Melzani G, Mori G, et al. Relapses after treatment of external genital warts are more frequent in HIV-positive patients than in HIV-negative controls. Sex Transm Dis 2002;29:121--5.
- Silverberg MJ, Ahdieh L, Munoz A, et al. The impact of HIV infection and immunodeficiency on human papillomavirus type 6 or 11 infection and on genital warts. Sex Transm Dis 2002;29:427--35.
- Bienvenu B, Martinez F, Devergie A, et al. Topical use of cidofovir induced acute renal failure. Transplantation 2002;73:661--2.
- King MD, Reznik DA, O'Daniels CM, et al. Human papillomavirus-associated oral warts among human immunodeficiency virus-seropositive patients in the era of highly active antiretroviral therapy: an emerging infection. Clin Infect Dis 2002;34:641--8.
- Hodgson TA, Greenspan D, Greenspan JS. Oral lesions of HIV disease and HAART in industrialized countries. Adv Dent Res 2006;19:57--62.
- Baccaglini L, Atkinson JC, Patton LL, et al. Management of oral lesions in HIV-positive patients. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2007;103:e1--23.
- Reeves W, Ruparelia SS, Swanson KI, et al. National registry for juvenile-onset recurrent respiratory papillomatosis. Arch Otolaryngol Head Neck Surg 2003;129:976--82.
- Moscicki AB, Shiboski S, Hills NK, et al. Regression of low-grade squamous intra-epithelial lesions in young women. Lancet 2004;364:1678--83.
- Wright TC, Koulos J, Schnoll F, et al. Cervical intraepithelial neoplasia in women infected with the human immunodeficiency virus: outcome after loop electrosurgical excision. Gynecol Oncol 1994;55:253--8.
- Fruchter RG, Maiman M, Sedlis A, et al. Multiple recurrences of cervical intraepithelial neoplasia in women with the human immunodeficiency virus. Obstet Gynecol 1996;87:338--44.
- Goldie SJ, Kuntz KM, Weinstein MC, et al. The clinical effectiveness of screening for anal squamous intraepithelial lesionsi homosexual and bisexual HIV-positive men. JAMA 1999;281:1822--9.
- Kurman RJ, Henson DE, Herbst AL, et al. Interim guidelines for management of abnormal cervical cytology. The 1992 National Cancer Institute Workshop. JAMA 1994;271:1866--9.
- Maiman M, Watts DH, Andersen J, et al. Vaginal 5-fluorouracil for high-grade cervical dysplasia in human immunodeficiency virus infection: a randomized trial. Obstet Gynecol 1999;94:954--61.
- Astrom KE, Mancall EL, Richardson EP J. Progressive multifocal leuko-encephalopathy; a hitherto unrecognized complication of chronic lymphatic leukaemia and Hodgkin's disease. Brain 1958;81:93--111.
- Stolt A., Sasnauskas K, Koskela P, et al. Seroepidemiology of the human polyomaviruses. J Gen Virol 2003;84(Pt 6):1499--504.
- Rodrigues C, Pinto D, Medeiros R. Molecular epidemiology characterization of the urinary excretion of polyomavirus in healthy individuals from Portugal---a Southern European population. J Med Virol 2007;79:1194--8.
- Gu ZY, Li Q, Si YL, et al. Prevalence of BK virus and JC virus in peripheral blood leukocytes and normal arterial walls in healthy individuals in China. J Med Virol 2003;70:600--5.
- Ciuta ST, Boros S, Napoli PA, et al. Predictors of survival in children with acquired immunodeficiency syndrome in Italy, 1983 to 1995. AIDS Patient Care STDS 1998;12:629--37.
- Araujo AP, Pereira HS, Oliveira RH, et al. Progressive multifocal leukoencephalopathy in a child with acquired immunodeficiency syndrome (AIDS). Arg Neuropsiguiatr 1997;55:122--5.
- Robinson LG, Chiriboga CA, Champion SE, et al. Progressive multifocal leukoencephalopathy successfully treated with highly active antiretroviral therapy and cidofovir in an adolescent infected with perinatal human immunodeficiency virus (HIV). J Child Neurol 2004;19:35--8.
- Wilmshurst JM, Burgess J, Hartley P, et al. Specific neurologic complications of human immunodeficiency virus type 1 (HIV-1) infection in children. J Child Neurol 2006;21:788--94.
- Shah I, Chudgar P. Progressive multifocal leukoencephalopathy (PML) presenting as intractable dystonia in an HIV-infected child. J Trop Pediatr 2005;51:380--2.
- Berger JR, Scott G, Albrecht J, et al. Progressive multifocal leukoencephalopathy in HIV-1-infected children. AIDS 1992;6:837--41.
- Liptai Z, Papp E, Barsi P, et al. Progressive multifocal leukoencephalopathy in an HIV-infected child. Neuropediatrics 2007;38:32--5.
- Angelini L, Pietrogrande MC, Delle Piane MR, et al. Progressive multifocal leukoencephalopathy in a child with hyperimmunoglobulin E recurrent infection syndrome and review of the literature. Neuropediatrics 2001;32:250--5.
- Antinori A, Cingolani A, Lorenzini P, et al. Clinical epidemiology and survival of progressive multifocal leukoencephalopathy in the era of highly active antiretroviral therapy: data from the Italian Registry Investigative Neuro AIDS (IRINA). J Neurovirol 2003;9(Suppl 1):47--53.
- Koralnik IJ. New insights into progressive multifocal leukoencephalopathy. Curr Opin Neurol 2004;17:365--70.
- Berenguer JP, Miralles P, Arrizabalaga J, et al. Clinical course and prognostic factors of progressive multifocal leukoencephalopathy in patients treated with highly active antiretroviral therapy. Clin Infect Dis 2003;36:1047--52.
- Mamidi A, DeSimone JA, Pomerantz RJ. Central nervous system infections in individuals with HIV-1 infection. J Neurovirol 2002;8:158--67.
- Bossolasco S, Calori G, Moretti F, et al. Prognostic significance of JC virus DNA levels in cerebrospinal fluid of patients with HIV-associated progressive multifocal leukoencephalopathy. Clin Infect Dis 2005;40:738--44.
- Drake AK, Loy CT, Brew BJ, et al. Human immunodeficiency virus-associated progressive multifocal leucoencephalopathy: epidemiology and predictive factors for prolonged survival. Eur J Neurol 2007;14:418--23.
- Hall CD, Dafni U, Simpson D, et al. Failure of cytarabine in progressive multifocal leukoencephalopathy associated with human immunodeficiency virus infection. AIDS Clinical Trials Group 243 Team. N Engl J Med 1998;338:1345--51.
- Marra CM, Rajicic N, Barker DE, et al. A pilot study of cidofovir for progressive multifocal leukoencephalopathy in AIDS. AIDS 2002;16:1791--7.
- Geschwind MD, Skolasky RI, Royal WS, et al. The relative contributions of HAART and alpha-interferon for therapy of progressive multifocal leukoencephalopathy in AIDS. J Neurovirol 2001;7:353--7.
- Giudici B, Vaz B, Bossolasco S, et al. Highly active antiretroviral therapy and progressive multifocal leukoencephalopathy: effects on cerebrospinal fluid markers of JC virus replication and immune response. Clin Infect Dis 2000;30:95--9.
- Safdar A, Rubocki RJ, Horvath JA, et al. Fatal immune restoration disease in human immunodeficiency virus type 1-infected patients with progressive multifocal leukoencephalopathy: impact of antiretroviral therapy-associated immune reconstitution. Clin Infect Dis 2002;35:1250--7.
- Cinque P, Koralnik IJ, Clifford DB. The evolving face of human immunodeficiency virus-related progressive multifocal leukoencephalopathy: defining a consensus terminology. J Neurovirol 2003;9(Suppl 1):88--92.
- D'Amico R, Sarkar S, Yusuff J, et al. Immune reconstitution after potent antiretroviral therapy in AIDS patients with progressive multifocal leukoencephalopathy. Scand J Infect Dis 2007;39:347--50.
- Du Pasquier RA, Koralnik IJ. Inflammatory reaction in progressive multifocal leukoencephalopathy: harmful or beneficial? J Neurovirol 2003;9(Suppl 1):25--31.
- Gershon A, Seward J, Takahashi M. Varicella vaccine. In: Plotkin S, Orenstein W, Offit PA. Vaccine. 5th ed. Philadelphia, PA: Saunders;2008:915--958.
- Gershon A, Mervish N, LaRussa P, et al. Varicella-zoster virus infection in children with underlying human immunodeficiency virus infection. J Infect Dis 1997;176:1496--500.
- Derryck A, LaRussa P, Steinberg S, et al. Varicella and zoster in children with human immunodeficiency virus infection. Pediatr Infect Dis J 1998;17:931--3.
- Hambelton S, Gershon AA. Preventing varicella-zoster disease. Clin Microbiol Rev 2005;18:70--80.
- LaRussa P, Steinberg SP, Seeman MD, et al. Determination of immunity to varicella-zoster virus by means of an intradermal skin test. J Infect Dis 1985;152:869--75.
- Veenstra J, Krol A, van Praag RM, et al. Herpes zoster, immunological deterioration and disease progression in HIV-1 infection. AIDS 1995;9:1153--8.
- Chen JJ, Zhu Z, Gershon AA, et al. Mannose 6-phosphate receptor dependence of varicella zoster virus infection in vitro and in the epidermis during varicella and zoster. Cell 2004;119:915--26.
- Ross AH. Modification of chicken pox in family contacts by administration of gamma globulin. N Engl J Med 1962;267:369--76.
- Clark R, Wilson S, Williams T. Varicella immunity in women infected with the human immunodeficiency virus. Clin Infect Dis 1994;19:1165--6.
- Schulze A, Dietzsch HJ. The natural history of varicella embryopathy: a 25-year follow-up. J Pediatr 2000;137:871--4.
- Arvin AM. Varicella-zoster virus. Clin Microbiol Reb 1996:9:361--81.
- Pastuszak AL, Levy M, Schick B, et al. Outcome after maternal varicella infection in the first 20 weeks of pregnancy. N Engl J Med 1994;330:901--5.
- Ussery XT, Annunziato P, Gershon AA, et al. Congenital varicella-zoster virus infection and Barrett's esophagus. J Infect Dis 1998;178:539--43.
- von Seidlein L, Gillette SG, Bryson Y, et al. Frequent recurrence and persistence of varicella-zoster virus infections in children infected with human immunodeficiency virus type 1. J Pediatr 1996;128:52--7.
- Martinez E, Gatell J, Morán Y, et al. High incidence of herpes zoster in patients with AIDS soon after therapy with protease inhibitors. Clin Infect Dis 1998;27:1510--3.
- Jura E, Chadwick EG, Josephs SH, et al. Varicella-zoster virus infections in children infected with human immunodeficiency virus. Pediatr Infect Dis J 1989;8:586--90.
- Kelley R, Mancao M, Lee F, et al. Varicella in children with perinatally acquired human immunodeficiency virus infection. J Pediatr 1994;124:271--3.
- Leibovitz E, Cooper D, Giurgiutiu D, et al. Varicella-zoster virus infection in Romanian children infected with the human immunodeficiency virus. Pediatrics 1993;92:838--42.
- Leibovitz E, Kaul A, Rigaud M, et al. Chronic varicella zoster in a child infected with human immunodeficiency virus: case report and review of the literature. Cutis 1992;49:27--31.
- Pahwa S, Biron K, Lim W, et al. Continuous varicella-zoster infection associated with acyclovir resistance in a child with AIDS. JAMA 1988;260:2879--82.
- Silliman CC, Tedder D, Ogle JW, et al. Unsuspected varicella-zoster virus encephalitis in a child with acquired immunodeficiency syndrome. J Pediatr 1993;123:418--22.
- Gershon A, Chen J, LaRussa P, et al. Varicella-zoster virus. In: Murray PR, Baron EJ, Jorgenson JH, Pfaller MA, Yolken RH, eds. Manual of clinical microbiology. 8th ed. Washington, DC: ASM Press: 2007;1319--30.
- Purdy KW, Heckenlively JR, Church JA, et al. Progressive outer retinal necrosis caused by varicella-zoster virus in children with acquired immunodeficiency syndrome. Pediatr Infect Dis J 2003;22:384--6.
- Hellinger WC, Bolling JP, Smith TF, et al. Varicella-zoster virus retinitis in a patient with AIDS-related complex: case report and brief review of the acute retinal necrosis syndrome. Clin Infect Dis 1993;16:208--12.
- Engstrom RE, Jr, Holland GN, Margolis TP, et al. The progressive outer retinal necrosis syndrome. A variant of necrotizing herpetic retinopathy in patients with AIDS. Ophthalmology 1994;101:1488--502.
- Franco-Paredes C, Bellehemeur T, Merchant A, et al. Aseptic meningitis and optic neuritis preceding varicella-zoster progressive outer retinal necrosis in a patient with AIDS. AIDS 2002;16:1045--9.
- Levin MJ, Dahl KM, Weinberg A, et al. Development of resistance to acyclovir during chronic infection with the Oka vaccine strain of varicella-zoster virus, in an immunosuppressed child. J Infect Dis 2003;188:954--9.
- Levin MJ, Gershon AA, Weinberg A, et al. Immunization of HIV-infected children with varicella vaccine. J Pediatr 2001;139:305--10.
- Levin MJ, Gershon AA, Weinberg A, et al. Administration of live varicella vaccine to HIV-infected children with current or past significant depression of CD4(+) T cells. J Infect Dis 2006;194:247--55.
- Bekker V, Westerlaken GH, Scherpbier H, et al. Varicella vaccination in HIV-1-infected children after immune reconstitution. AIDS 2006;20:2321--9.
- Sharrar RG, LaRussa P, Galea SA, et al. The postmarketing safety profile of varicella vaccine. Vaccine 2000;19:916--23.
- Kramer JM, LaRussa P, Tsai WC, et al. Disseminated vaccine strain varicella as the acquired immunodeficiency syndrome-defining illness in a previously undiagnosed child. Pediatrics 2001;108:E39.
- CDC. A new product (VariZIG) for postexposure prophylaxis of varicella available under an investigational new drug application expanded access protocol. MMWR 2006;55:209--10.
- Asano Y, Yoshikawa T, Suga S, et al. Postexposure prophylaxis of varicella in family contact by oral acyclovir. Pediatrics 1993;92:219--22.
- Huang YC, Lin TY, CH C. Acyclovir prophylaxis of varicella after household exposure. Pediatr Infect Dis J 1995;14:152--4.
- Lin TY, Huang YC, Ning HC, et al. Oral acyclovir prophylaxis of varicella after intimate contact. Pediatr Infect Dis J 1997;16:1162--5.
- Balfour HH, Jr, Bean B, Laskin OL, et al. Acyclovir halts progression of herpes zoster in immunocompromised patients. N Engl J Med 1983;308:1448--53.
- Yin PD, Kurup SK, Fischer SH, et al. Progressive outer retinal necrosis in the era of highly active antiretroviral therapy: successful management with intravitreal injections and monitoring with quantitative PCR. J Clin Virol 2007;38:254--9.
- Austin RB. Progressive outer retinal necrosis syndrome: a comprehensive review of its clinical presentation, relationship to immune system status, and management. Clin Eye Vis Care 2000;12:119--29.
- Dworkin RH, Johnson RW, Breuer J, et al. Recommendations for the management of herpes zoster. Clin Infect Dis 2007;44(Suppl 1):S1--26.
- Dekker CL, Prober CG. Pediatric uses of valacyclovir, penciclovir and famciclovir. Pediatr Infect Dis J 2001;20:1079--81.
- Domingo P, Torres OH, Ris J, et al. Herpes zoster as an immune reconstitution disease after initiation of combination antiretroviral therapy in patients with human immunodeficiency virus type-1 infection. Am J Med 2001;110:605--9.
- Heininger U, Seward JF. Varicella. Lancet 2006;368:1365--76.
- Enright A, Prober C. Antiviral therapy in children with varicella zoster virus and herpes simplex virus infections. Herpes 2003;10:32--7.