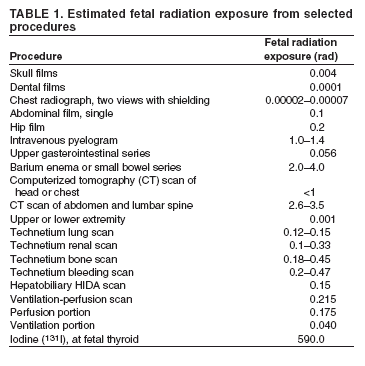
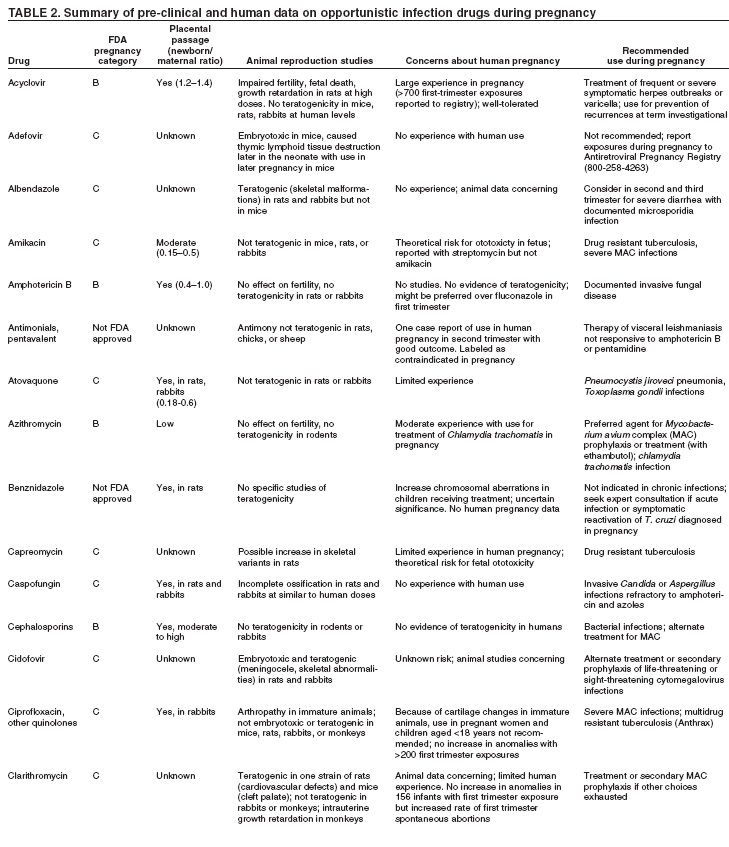
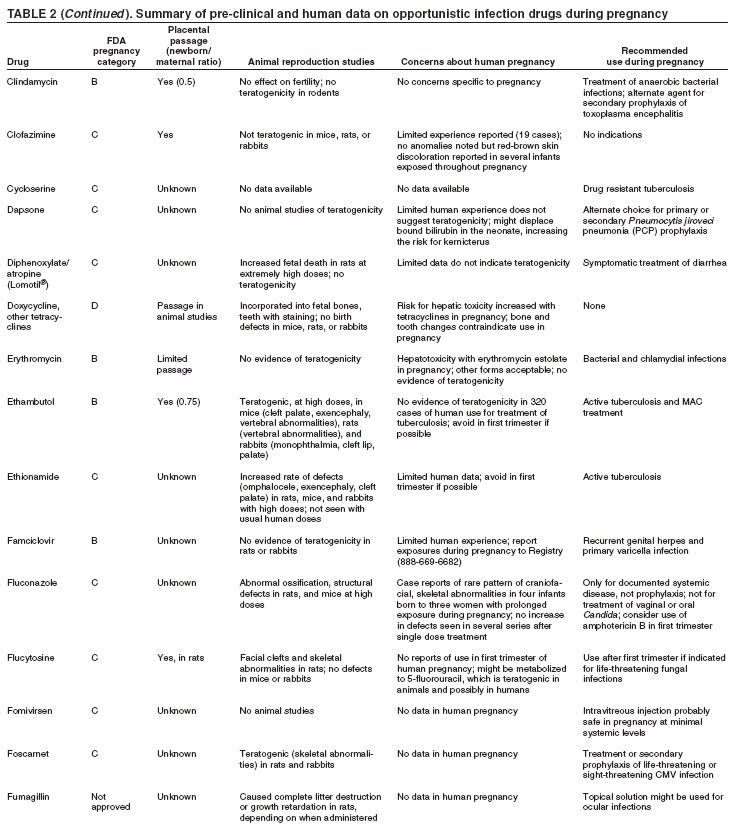
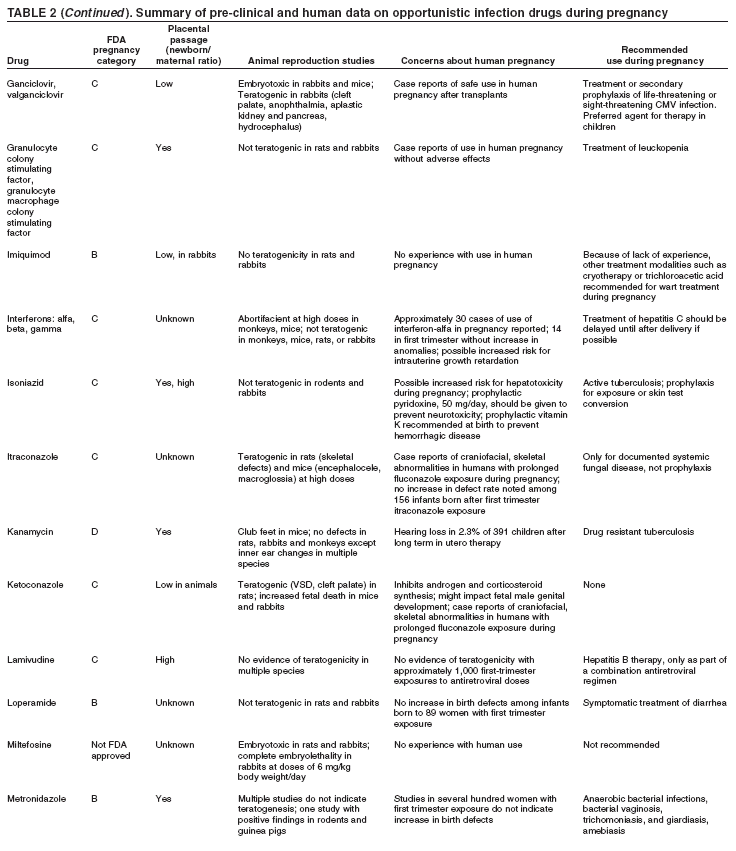
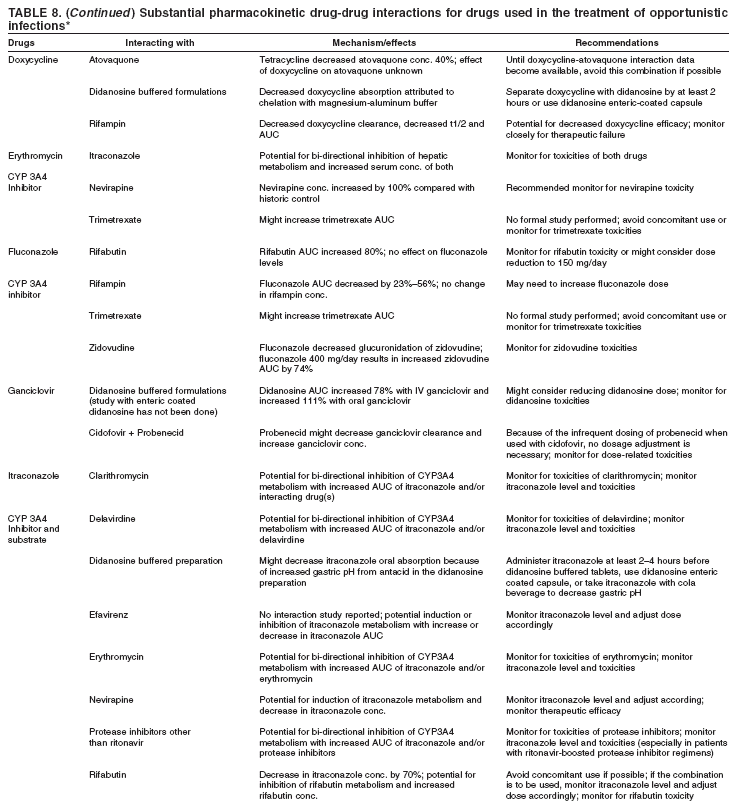
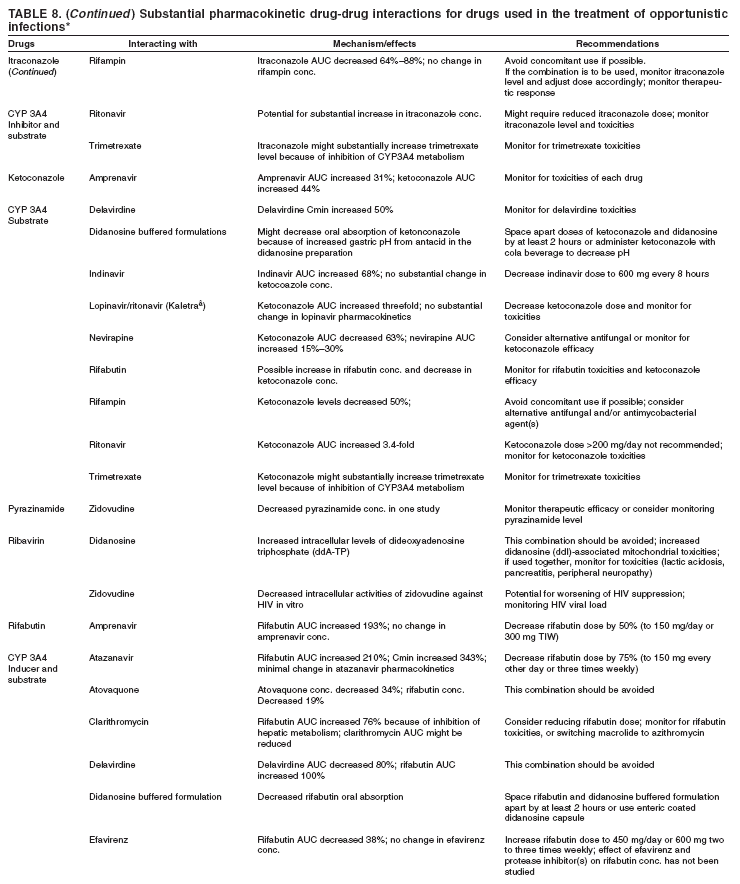
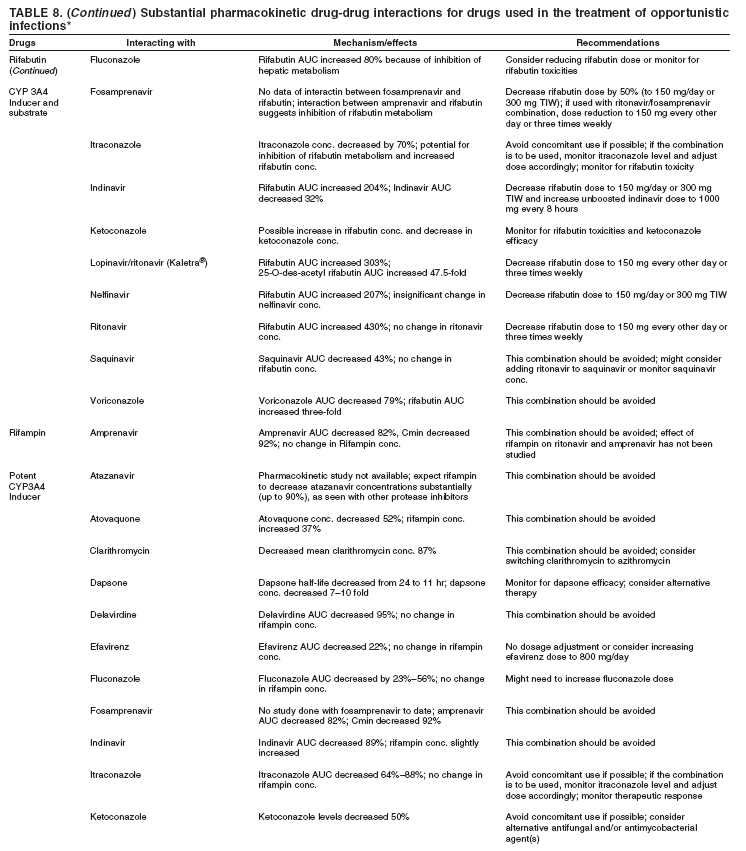
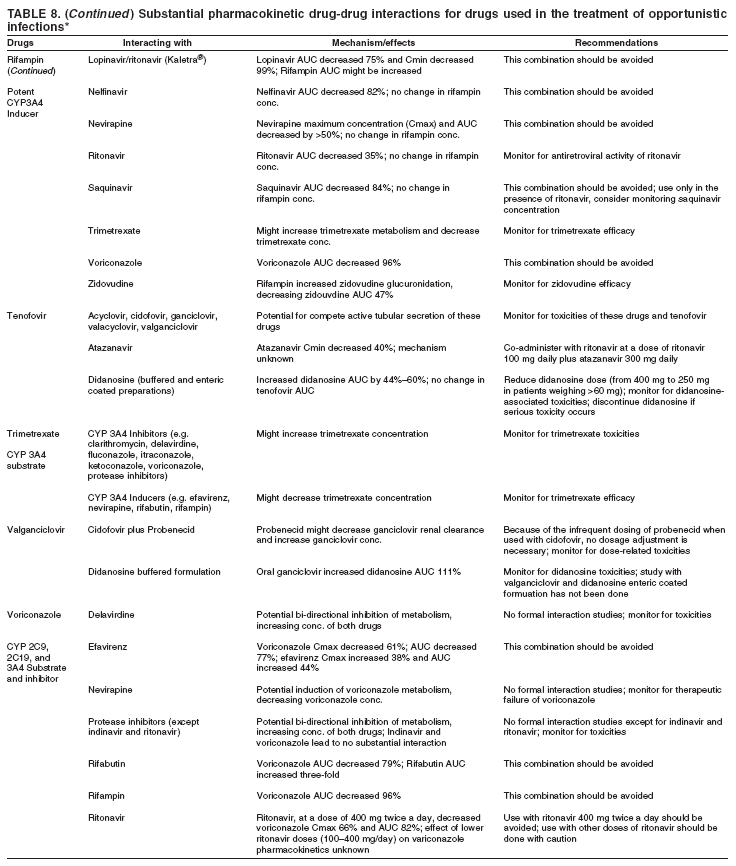
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
| ||||||||||
|
|
|
|
|
Persons using assistive technology might not be able to fully access information in this file. For assistance, please send e-mail to: mmwrq@cdc.gov. Type 508 Accommodation and the title of the report in the subject line of e-mail. Treating Opportunistic Infections Among HIV-Infected Adults and AdolescentsRecommendations from CDC, the National Institutes of Health, and the HIV Medicine Association/Infectious Diseases Society of AmericaAn erratum has been published for this article. To view the erratum, please click here. Prepared by The material in this report originated in the Office of the Director, National Center for HIV, STD and TB Prevention, Janet L. Collins, M.D., Acting Director. Corresponding Author: Constance A. Benson, M.D., Antiviral Research Center, University of California, San Diego, 150 W. Washington St., Suite 100, San Diego, CA 92103. Telephone: 619-543-8080; Fax: 619-298-0177; e-mail: cbenson@ucsd.edu. Summary The National Institutes of Health, the HIV Medicine Association of the Infectious Diseases Society of America, and CDC have developed guidelines for treatment of opportunistic infections (OIs) among adults and adolescents infected with human immunodeficiency virus (HIV). These guidelines are intended for clinicians and other health-care providers who care for HIV-infected adults and adolescents, including pregnant women; they complement companion guidelines for treatment of OIs among HIV-infected children and previously published guidelines for prevention of OIs in these populations. They include evidence-based guidelines for treatment of 28 OIs caused by protozoa, bacteria, fungi, and viruses, including certain OIs endemic in other parts of the world but that might be observed in patients in the United States. Each OI section includes information on epidemiology, clinical manifestations, diagnosis, treatment recommendations, monitoring and adverse events, management of treatment failure, prevention of recurrence, and special considerations in pregnancy. Tables address drugs and doses, drug toxicities, drug interactions, adjustment of drug doses in persons with reduced renal function, and data about use of drugs in pregnant women. IntroductionOpportunistic infections (OIs) continue to cause morbidity and mortality in patients with human immunodeficiency virus (HIV)-1 infection throughout the world. Potent combination antiretroviral therapy (ART) has reduced the incidence of OIs for certain patients with access to care. However, certain patients in the developed and developing world do not have access to care and have OIs. Other patients do not have a sustained response to antiretroviral agents for multiple reasons, including poor adherence, drug toxicities, drug interactions, or initial acquisition of a drug-resistant strain of HIV-1. Therefore, OIs will continue to cause substantial morbidity and mortality in patients with HIV-1 infection. The therapy of OIs has changed substantially during the AIDS epidemic. As more information about efficacy, toxicity, and interactions of the drugs to treat and prevent OIs has emerged, management strategies have evolved. New drugs have also become available that occupy important roles in our therapeutic armamentarium. These guidelines and the accompanying guidelines, Treating Opportunistic Infections Among HIV-Exposed and Infected Children, join two previous guidelines, The United States Public Health Service-Infectious Diseases Society of America Guidelines for the Prevention of Opportunistic Infections in Persons Infected with the Human Immunodeficiency Virus and The Department of Health and Human Services (DHHS) Guidelines for the Use of Antiretroviral Agents in HIV-Infected Adults and Adolescents. The current guidelines share key features with their companion guidelines:
How To Use the Information in This ReportFor each of the diseases covered in this report, specific recommendations are provided. Recommendations are rated by the IDSA rating system. In this system, the letters A through E signify the strength of the recommendation for or against a treatment measure, and Roman numerals I through III indicate the quality of evidence supporting the recommendation (Box). Effect of Antiretroviral Therapy on the Incidence and Management of OIsData from both randomized controlled trials and observational cohort studies document that antiretroviral therapy (ART) reduces the incidence of OIs and improves survival, independent of the use of antimicrobial prophylaxis, and reduces overall mortality among persons with HIV-1 infection (1--7). Potent ART does not replace the need for antimicrobial prophylaxis among patients with severe immune suppression. However, ART is the cornerstone of the overall strategy to reduce morbidity attributed to HIV-1--related infections and other HIV1--related processes. The clinical benefit of ART in reducing the risk for OIs over the short term has been best demonstrated for those with a CD4+ T lymphocyte count <200 cells/µL. Studies also support benefit in patients with CD4+ T lymphocyte counts >200 cells/µL, although the overall benefit of starting ART in this population is uncertain. Improvements in specific measures of immune function, including pathogen-specific immunity, have been well documented among patients who initiated ART at CD4+ T lymphocyte counts >200 cells/µL (8--10). Whether such measures correlate with clinical protection against infection or other HIV-1-related complications remains to be determined. In addition to preventing OIs, ART can lead to resolution or improvement of certain OIs, most notably for those where specific treatment is not available. Treatment of patients with ART in the setting of an OI also can result in an exuberant inflammatory reaction that might require the use of anti-inflammatory agents for clinical management. Finally, patients who receive potent ART can have atypical presentations of OIs either early after the initiation of ART or after prolonged treatment. Specific guidelines for the management of ART in the presence of acute OIs have not previously been developed. Two principal circumstances to consider include the initiation of ART in the setting of an acute OI, and the management of ART when an acute OI occurs in a patient who is already receiving ART. The management in each circumstance will vary depending on the degree of virologic and immunologic disease progression before initiation of ART and the virologic and immunologic benefit resulting from ART, the duration of HIV-1 disease before and since starting ART, and the potential for drug-drug interactions between the ART regimen and the treatment needed for the OI. Initiation of ART in the Setting of an Acute OI (Treatment-Naïve Patients)The benefits of ART in the setting of an acute OI include the improvement in immune function that would potentially contribute to faster resolution of the OI. The beneficial effect of initiating ART during an acute OI has been best demonstrated for OIs for which limited or no effective therapies are available. Reports detailing the resolution of cryptosporidiosis, microsporidiosis, progressive multifocal leukoencephalopathy (PML), and Kaposi sarcoma after the initiation of potent ART provide evidence that improving immune function can lead to improved outcome in the setting of an acute OI (11--14). Another benefit of immediate initiation of potent ART during an acute OI is the reduction in risk for a second OI. Arguments against the immediate initiation of ART concurrent with the diagnosis of an OI include drug toxicities including additive toxicities, distinguishing toxicities caused by antiretrovirals (ARVs) from toxicities related to drugs used to manage OIs, the potential for drug interactions between OI therapies and ART, and the potential for inflammatory immune reconstitution syndromes to complicate the management of the OI in this setting. Much simpler ART regimens are available for the treatment of HIV-1 disease, diminishing the argument to delay therapy for reasons of complexity. However, overlapping toxicities exist between OI treatments and ART regimens that can complicate the ability to identify drug specific toxicity. Drug interactions pose the biggest problem for the treatment of patients with tuberculosis (TB), but ART regimens compatible with TB treatment are available. Immune reconstitution syndromes have been described for mycobacterial infections (including disease caused by Mycobacterium avium complex [MAC] and Mycobacterium tuberculosis, Pneumocystis jiroveci pneumonia (PCP), toxoplasmosis, hepatitis B and hepatitis C viruses, cytomegalovirus (CMV) infection, varicella-zoster virus (VZV) infection, cryptococcal infection and PML (12,15--25). Immune reconstitution syndromes are characterized by fever and worsening of the clinical manifestations of the OI or new manifestations weeks after the initiation of ART. Determining the absence of recrudescence of the underlying OI and new drug toxicity or a new OI is important. If the syndrome does represent an immune reactivation syndrome, adding nonsteroidal anti-inflammatory agents or corticosteroids to alleviate the inflammatory reaction is appropriate. The inflammation might take weeks or months to subside. The largest number of published reports of immune reconstitution syndromes is among patients with TB disease. Patients can experience high fevers, worsening lymphadenopathy or transient-to-severe worsening of pulmonary infiltrates, and expanding central nervous system lesions (19,26,27). Such "paradoxical reactions" might be more common among HIV-1--infected patients with TB disease who were started on potent ART compared with those not started on ART and among patients with TB disease who were not HIV-1-infected (19). Reduction of HIV-1 RNA levels and marked increases in CD4+ T lymphocyte counts have been associated with the occurrence of paradoxical reactions in patients with TB disease or MAC (15,17,19, 26). Although the majority of reactions occur within the first few weeks after initiation of ART, some have occurred up to several months after the initiation of TB therapy or ART. No randomized controlled trials exist that demonstrate that initiating ART improves outcome for patients being treated with specific therapy for their acute OI. In addition, no data demonstrate that initiation of ART in the setting of an acute OI worsens the prognosis or treatment for that OI. Trials are underway to evaluate the most appropriate timing for initiation of ART in this context. Management of Acute OIs in the Setting of ARTOIs that develop after patients have been started on potent ART can be categorized into three groups. The first group includes OIs that occur shortly after initiating ART (within 12 weeks). These cases are thought to be subclinical infections that have been unmasked by early immune reconstitution and are not considered to be early failure of ART (10,15,17,28--31). The second group includes reports of OIs occurring >12 weeks after initiation of ART among patients with suppressed HIV-1 RNA levels and sustained CD4+ T lymphocyte counts >200 cells/µL (32,33). Two cases of spinal MAC among patients with nadir CD4+ T lymphocyte counts <50 cells/µL who had sustained CD4+ T lymphocyte count increases to >200 cells/µL are examples. Determining whether these represent a form of immune reconstitution syndrome as opposed to incomplete immunity with the occurrence of a new OI is difficult. The presence of organisms by stain and culture suggests that, in either situation, specific therapy is indicated. The third group includes OIs that develop among patients who are experiencing virologic and immunologic failure while on potent ART. These represent clinical failure of ART. When To Initiate ART in the Setting of an OINo consensus has been reached about the optimal time to start ART in the presence of a recently diagnosed OI. The decision to start potent ART should take into consideration the availability of effective therapy for the OI, the risk for drug interactions, overlapping drug toxicities, the risk for and consequences of the development of an inflammatory immune reconstitution syndrome, and the willingness and ability of patients to take and adhere to their regimens. In cases of cryptosporidiosis, microsporidiosis, PML, and Kaposi sarcoma, the early benefits of potent ART outweigh any increased risk, and potent ART should be started as soon as possible (AIII). In the setting of TB disease, MAC, PCP, and cryptococcal meningitis, awaiting a response to OI therapy is usually warranted before initiating ART (CIII). When an OI occurs within 12 weeks of starting ART, treatment for the OI should be started, and ART should be continued (AIII). When an OI occurs despite complete virologic suppression (i.e., late OI), therapy for the OI should be initiated, potent ART should be continued, and if the CD4+ T cell response to ART has been suboptimal, modification of the ART regimen may be considered (CIII). When an OI occurs in the setting of virologic failure, OI therapy should be started, antiretroviral resistance testing should be performed, and the ART regimen should be modified if possible to achieve better virologic control (AI). Special Considerations During PregnancyNo large studies have been conducted on the epidemiology or manifestations of HIV-1--associated OIs among pregnant women. No data demonstrate that the spectrum differs from that among nonpregnant women with comparable CD4+ T lymphocyte counts. CD4+ T lymphocyte counts characteristically drop during pregnancy, probably because of dilutional effects of the increased plasma volume. CD4+ T lymphocyte percentages are generally more stable and should be used for determining degree of immune suppression during pregnancy (34--36). Physiologic changes occur during pregnancy that might impact the presentation of acute OIs and the considerations for implementing OI treatment or antiretroviral therapies. These changes include (37):
Pregnancy also impacts decisions about diagnostic testing. Fetal risk is not increased with cumulative radiation doses below 5 rads. Teratogenesis is observed in animals at doses of 100--200 rads. In humans, the primary risk associated with high dose radiation exposure is growth restriction, microcephaly, and developmental disabilities. The most vulnerable period is 8--15 menstrual weeks of gestation with minimal risk before 8 weeks and after 25 weeks. The apparent threshold for development of mental retardation is 20--40 rads, with risk increasing linearly with increasing exposures above this level. Among children, risk for carcinogenesis might be increased approximately one per 1,000 or less per rad of in utero radiation exposure (38). The majority of radiographic and nuclear medicine studies result in radiation exposure to the fetus that is much lower than the 5 rad recommended limit; therefore, pregnancy should not preclude usual diagnostic evaluation when an OI is suspected (Table 1) (38--40). Abdominal shielding should be used when feasible to further limit radiation exposure to the fetus. Experience with use of magnetic resonance imaging (MRI) in pregnancy is limited. Although no adverse fetal effects have been reported, the National Radiological Protection Board advises against use of MRI in the first trimester (38). Other procedures necessary for diagnosis of suspected OIs should be performed in pregnancy as indicated for nonpregnant patients. Pregnant women who are >20 weeks of gestation should not lie flat on their backs but should have the left hip elevated with a wedge to displace the uterus off of the great vessels and prevent supine hypotension. Adequate oxygenation should be maintained. Because of the serious nature of OIs among HIV-1--infected persons, diagnostic procedures and indicated therapy should not be withheld during pregnancy; the therapy with the least potential toxicity should be selected (Table 2). The predictive value of animal data for effects in humans is unclear. In addition, reproductive studies among animals usually include only one drug at a time, and HIV-1--infected pregnant women might be using multiple antiretroviral, OI, and other drugs concurrently. The potential for enhanced toxicity of combinations of drugs has not been evaluated. For pregnant women who have had an OI diagnosed and are not on ART, immediate initiation of ART with OI therapy should be encouraged (AIII) (41). Decisions about immediate versus delayed initiation of ART in pregnancy should take into account gestational age, maternal HIV-1 RNA levels and clinical condition, and potential toxicities and interactions between ART and OI drugs. Pregnant women with active OIs who receive drugs for which information about their use in pregnancy is limited should have additional evaluation of fetal growth and well-being. After first trimester exposure to agents of uncertain teratogenic potential, a detailed ultrasound examination at 18--20 weeks should be conducted to detect major anomalies, although the ultrasound will not detect all anomalies. For women who receive drugs throughout pregnancy or in the third trimester for which information about their use in pregnancy is limited, an ultrasound should be conducted every 4--6 weeks to assess fetal growth and fluid volume. Pregnant women in the third trimester should be instructed in daily fetal movement counting to detect decreased activity that might indicate fetal compromise. Weekly fetal nonstress testing should be initiated at 32 weeks of gestation unless indicated sooner based on clinical or ultrasound findings (42). Disease Specific RecommendationsPneumocystis Jiroveci PneumoniaEpidemiology Pneumocystis jiroveci pneumonia (PCP) is caused by Pneumocystis jiroveci, a ubiquitous organism classified as a fungus but that shares biologic characteristics with protozoa. The taxonomy of the organism has been changed; Pneumocystis carinii now refers only to the pneumocystis that infects rodents, and Pneumocystis jiroveci refers to the distinct species that infects humans. The abbreviation PCP is still used to designate Pneumocystis pneumonia. Initial infection with P. jiroveci usually occurs in early childhood; two thirds of healthy children have antibody to P. jiroveci by age 2--4 years (43). PCP is a result either of reactivation of latent infection or new exposure to the organism. Rodent studies and case clusters among immunosuppressed patients indicate that spread among persons can occur by the airborne route. Disease probably occurs by new acquisition and by reactivation (44,45). Before the widespread use of primary PCP prophylaxis and effective ART, PCP occurred in 70%--80% of patients with AIDS (46). The course of treated PCP was associated with a mortality of 20%--40% in persons with profound immunosuppression. Approximately 90% of cases occurred among patients with CD4+ T lymphocyte counts of <200/µL. Other factors associated with a higher risk of PCP included CD4+ T lymphocyte percentage <15%, previous episodes of PCP, oral thrush, recurrent bacterial pneumonia, unintentional weight loss, and higher plasma HIV-1 RNA (47,48). Incidence of PCP has declined substantially with widespread use of prophylaxis and effective ART; recent incidence rates among patients with AIDS in Western Europe and the U.S. are 2--3 cases per 100 person-years (49). The majority of cases occur among patients who are unaware of their HIV-1 infection or are not receiving ongoing HIV care (50) or among those with advanced immunosuppression (CD4+ T lymphocyte counts <100 cells/µL) (51). Clinical Manifestations The most common manifestations of PCP among HIV-1--infected persons are the subacute onset of progressive exertional dyspnea, fever, nonproductive cough, and chest discomfort that worsens over a period of days to weeks. The fulminant pneumonia observed among non-HIV-1--infected patients is less common (52,53). In mild cases, pulmonary examination is usually normal at rest. With exertion, tachypnea, tachycardia, and diffuse dry ("cellophane") rales might be observed (53). Oral thrush is a common co-infection. Fever is apparent in the majority of cases and might be the predominant symptom among some patients. Extrapulmonary disease is rare but can present in any organ and has been associated with use of aerosolized pentamidine prophylaxis. Hypoxemia, the most characteristic laboratory abnormality, might range from mild-to-moderate (room air arterial oxygen [pO2] of >70 mm/Hg or alveolar-arterial O2 difference, [A-a] DO2 <35 mm/Hg) to moderate-to-severe levels (pO2 <70 mm/Hg or [A-a] DO2 >35 mm/Hg). Oxygen desaturation with exercise is indicative of an abnormal A-a gradient but is nonspecific (54). Elevation of lactate dehydrogenase levels to >500 mg/dL is common but nonspecific (55). The chest radiograph typically demonstrates diffuse, bilateral, symmetrical interstitial infiltrates emanating from the hiLa in a butterfly pattern (53); however, patients with early disease might have a normal chest radiograph (56). In addition, atypical presentations with nodules, asymmetric disease, blebs and cysts, upper lobe localization, and pneumothorax occur. Cavitation or pleural effusion is uncommon in the absence of other pulmonary pathogens or malignancy, and the presence of a pleural effusion might indicate an alternative diagnosis. Approximately 13%--18% of patients with documented PCP have another concurrent cause of pulmonary dysfunction (e.g., TB, Kaposi sarcoma, or bacterial pneumonia) (57,58). Pneumothorax in a patient with HIV-1 infection should raise the suspicion of PCP (59,60). Thin-section computerized tomography (CT) demonstrating patchy ground-glass attenuation (61) or a gallium scan showing increased pulmonary uptake (62) increases the likelihood that a diagnostic study such as bronchoscopy would demonstrate PCP in patients with mild-to-moderate symptoms and a normal chest radiograph and might be useful in adjunctive studies. However, a negative thin-section CT scan does not rule out PCP. Diagnosis Because the clinical presentation, blood tests, or chest radiographs are not pathognomonic for PCP and the organism cannot be routinely cultivated, histopathologic demonstration of organisms in tissue, bronchoalveolar lavage fluid, or induced sputum (57,58,63,64) samples is required for a definitive diagnosis. Spontaneously expectorated sputum has low sensitivity and should not be submitted to the laboratory to diagnose PCP. Cresyl violet, Giemsa, Diff-Quik, and Wright stains detect both the cyst and trophozoite forms but do not stain the cyst wall; Gomori Methenamine Silver, Gram-Weigert and toluidine blue stain the cyst wall. Certain laboratories prefer direct immunofluorescent staining. Nucleic acid tests are being developed, but their use remains experimental (65,66). Previous studies of stained respiratory tract samples obtained by various methods indicate the following relative diagnostic sensitivities: induced sputum <50 to >90% (the sensitivity and specificity depends heavily on the quality of the specimens and the experience of the microbiologist or pathologist), bronchoscopy with bronchoalveolar lavage 90%--99%, transbronchial biopsy 95%--100%, and open lung biopsy 95%--100%. Because of the potential for certain processes to have similar clinical manifestations, a specific diagnosis of PCP should be sought rather than relying on a presumptive diagnosis. Treatment can be initiated before making a definitive diagnosis because organisms persist in clinical specimens for days or weeks after effective therapy is initiated (64). Treatment Recommendations Trimethoprim-sulfamethoxazole (TMP-SMX) is the treatment of choice (67,68) (AI). The dose must be adjusted for abnormal renal function. Multiple randomized clinical trials indicate that TMP-SMX is as effective as parenteral pentamidine and more effective than other regimens. Adding leucovorin to prevent myelosuppression during acute treatment is not recommended because of questionable efficacy and some evidence for a higher failure rate (69) (DII). Oral outpatient therapy of TMP-SMX is highly effective among patients with mild-to-moderate disease (68) (AI). Mutations associated with resistance to sulfa drugs have been documented, but their effect on clinical outcome is uncertain (70). Patients who have PCP despite TMP-SMX prophylaxis are usually effectively treated with standard doses of TMPSMX (BIII). Patients with documented PCP and moderate-to-severe disease, as defined by room air pO2 <70 mm/Hg or arterial-alveolar O2 gradient >35 mm/Hg, should receive corticosteroids as early as possible, and certainly within 72 hours after starting specific PCP therapy (71--75) (AI). If steroids are started at a later time, their benefits are unclear, although the majority of clinicians would use them in such circumstances for patients with severe disease (BIII). The preferred corticosteroid dose and regimen is prednisone 40 mg by mouth twice a day for days 1--5, 40 mg daily for days 6--10, and 20 mg daily for days 11--21 (72,73) (AI). Methylprednisolone at 75% of the respective prednisone dose can be used if parenteral administration is necessary. Alternative therapeutic regimens include 1) dapsone and TMP for mild-to-moderate disease (69,76) (BI) (this regimen may have similar efficacy and fewer side effects than TMPSMX but is less convenient because of the number of pills); 2) primaquine plus clindamycin (77--79) (BI) (this regimen is also effective in mild-to-moderate disease, and the clindamycin component can be administered intravenously for more severe cases; however, primaquine is only available orally; 3) intravenous pentamidine (80--82) (AI) (generally the drug of second choice for severe disease); 4) atovaquone suspension (67,83) (BI) (this is less effective than TMP-SMX for mild-to-moderate disease but has fewer side effects); and 5) trimetrexate with leucovorin (84) (BI) (this is less effective than TMP-SMX but can be used if the latter is not tolerated and an intravenous regimen is needed). Leucovorin must be continued 3 days after the last trimetrexate dose. The addition of dapsone, sulfamethoxazole, or sulfadiazine to trimetrexate might improve efficacy on the basis of the sequential enzyme blockade of folate metabolism, although no study data exist to confirm this (CIII). Aerosolized pentamidine should not be used for the treatment of PCP because of limited efficacy and more frequent relapse (82,85,86) (DI). The recommended duration of therapy for PCP is 21 days (52) (AII). The probability and rate of response to therapy depends on the agent used, number of previous episodes, severity of illness, degree of immunodeficiency, and timing of initiation of therapy. Although the overall prognosis of patients whose degree of hypoxemia requires intensive care unit (ICU) admission or mechanical ventilation remains poor, survival in up to 40% of patients requiring ventilatory support has been reported in recent years (87--89). Because long-term survival is possible for patients in whom ART is effective, certain patients with AIDS and severe PCP should be offered ICU admission or mechanical ventilation when appropriate (e.g., when they have reasonable functional status) (AII). Because of the potential for additive or synergistic toxicities associated with anti-PCP and antiretroviral therapies, certain health-care providers delay initiation of ART until after the completion of anti-PCP therapy, despite some suggestion of potential benefit for early ART (88) (CIII). An immune recovery inflammatory syndrome has been described for PCP (90) and might complicate the concurrent administration of anti-PCP treatment and ART. Monitoring and Adverse Events Careful monitoring during therapy is important to evaluate response to treatment and to detect toxicity as soon as possible. Follow-up after therapy includes assessment for early relapse, especially when therapy has been with an agent other than TMP-SMX or was shortened for toxicity. PCP prophylaxis should be initiated promptly and maintained until the CD4+ T lymphocyte count is >200 cells/µL. If PCP occurred at a CD4+ T lymphocyte count >200 cells/µL, maintaining PCP prophylaxis for life regardless of the CD4+ T cell response might be prudent; however, data about the most appropriate approach in this setting are limited. Adverse reaction rates among patients with AIDS are high for TMP-SMX (20%--85%) (67,68,76,78,80,81,84,91--93). Common adverse effects are rash (30%--55%) (including Stevens-Johnson syndrome), fever (30%--40%), leukopenia (30%--40%), thrombocytopenia (15%), azotemia (1%--5%), hepatitis (20%), and hyperkalemia. Supportive care for common adverse effects should be attempted before discontinuing TMP-SMX (AIII). Rashes can often be "treated through" with antihistamines, nausea can be controlled with antiemetics, and fever can be managed with antipyretics. The most common adverse effects of alternative therapies include methemoglobinemia and hemolysis with dapsone or primaquine (especially in those with G-6-PD deficiency), rash, and fever with dapsone (68,76); azotemia, pancreatitis, hypo- or hyperglycemia, leukopenia, fever, electrolyte abnormalities, and cardiac dysrhythmia with pentamidine (80--83); anemia, rash, fever, diarrhea, and methemoglobinemia with primaquine and clindamycin (68,77,78); headache, nausea, diarrhea, rash, fever, and transaminase elevations with atovaquone (67,91); and bone marrow suppression, fever, rash, and hepatitis with trimetrexate (84). Management of Treatment Failure Clinical failure is defined by the lack of improvement or worsening of respiratory function documented by arterial blood gases after at least 4--8 days of anti-PCP treatment. Treatment failure attributed to treatment-limiting toxicities occurs in up to one third of patients (69). Failure attributed to lack of drug efficacy occurs in approximately 10% of those with mild-to-moderate disease. Adding or switching to another regimen is the appropriate management for treatment-related toxicity (BII). No convincing clinical trials exist to base recommendations for the management of treatment failure attributed to lack of drug efficacy. It is important to wait at least 4--8 days before switching therapy for lack of clinical improvement (BIII). In the absence of corticosteroid therapy, early and reversible deterioration within the first 3--5 days of therapy is typical, probably because of the inflammatory response caused by antibiotic-induced lysis of organisms in the lung. Other concomitant infections must be excluded as a cause for such deterioration (57,58). Bronchoscopy with bronchoalveolar lavage should be strongly considered even if it was conducted before initiating therapy. If TMP-SMX has failed or must be avoided for toxicity in moderate-to-severe disease, the common practice is to use parenteral pentamidine, primaquine combined with clindamycin, or trimetrexate (with or without oral dapsone) plus leucovorin (78,80,84) (BII). For mild disease, atovaquone is a reasonable alternative (BII). Although one meta-analysis concluded that the combination of clindamycin and primaquine might be the most effective regimen for salvage therapy (79), no prospective clinical trials have evaluated the optimal approach to patients who fail therapy with TMP-SMX. Prevention of Recurrence Patients who have a history of PCP should be administered secondary prophylaxis (chronic maintenance therapy) for life with TMP-SMX unless immune reconstitution occurs as a result of ART (94) (AI). For patients who are intolerant of TMP-SMX, alternatives are dapsone, dapsone combined with pyrimethamine, atovaquone, or aerosolized pentamidine. Secondary prophylaxis should be discontinued for adult and adolescent patients whose CD4+ T lymphocyte cell count has increased from <200 cells/µL to >200 cells/µL for at least 3 months as a result of ART (94--97) (AI). Secondary prophylaxis should be re-introduced if the CD4+ T lymphocyte count decreases to <200 cells/µL (AIII) or if PCP recurs at a CD4+ T lymphocyte count of >200 cells/µL (CIII). Special Considerations During Pregnancy Diagnostic considerations during pregnancy are the same as for nonpregnant women. Indications for therapy are the same as for nonpregnant women. The preferred initial therapy during pregnancy is TMP-SMX, although alternate therapies can be used if patients are unable to tolerate or are unresponsive to TMP-SMX (98) (AI). Neonatal care providers should be informed of maternal sulfa or dapsone therapy if used near delivery because of the theoretical increased risk for hyperbilirubinemia and kernicterus (99). Pentamidine is embryotoxic but not teratogenic among rats and rabbits (100). Trimetrexate should not be used because of teratogenicity at low doses in multiple animal studies, fetopathy in humans associated with use of the biochemically similar agents methotrexate and aminopterin, and the potential negative effects on placental and fetal growth (101) (EIII). Adjunctive corticosteroid therapy should be used as indicated in nonpregnant adults (102--105) (AIII). Maternal fasting and postprandial glucose levels should be monitored closely when corticosteroids are used in the third trimester because the risk for glucose intolerance is increased. Rates of preterm labor and preterm delivery are increased with pneumonia during pregnancy. Pregnant women with pneumonia after 20 weeks of gestation should be monitored for evidence of contractions (BII). Toxoplasma gondii EncephalitisToxoplasmic encephalitis (TE) is caused by the protozoan Toxoplasma gondii. Disease occurs almost exclusively because of reactivation of latent tissue cysts (106--109). Primary infection occasionally is associated with acute cerebral or disseminated disease. Seroprevalence varies substantially among different communities (e.g., approximately 15% in the United States and 50%--75% in certain European countries) (109,110). In the pre-ART era, for patients with advanced immunosuppression who were seropositive for T. gondii and not receiving prophylaxis with drugs active against T. gondii, the 12-month incidence of TE was approximately 33%. The incidence and associated mortality in Europe and the United States has decreased substantially with the initiation of ART and the broad use of prophylaxis regimens active against T. gondii (111--113). Clinical disease is rare among patients with CD4+ T lymphocyte counts >200 cells/µL. The greatest risk is among patients with a CD4+ T lymphocyte count <50 cells/µL (106--108). Primary infection occurs after eating undercooked meat containing tissue cysts or ingestion of oocysts that have been shed in cat feces and have sporulated in the environment (which requires at least 24 hours). No transmission of the organism occurs by person-to-person contact. Clinical Manifestations The most common clinical presentation of T. gondii infection among patients with AIDS is a focal encephalitis with headache, confusion, or motor weakness and fever (106--108). Physical examination might demonstrate focal neurological abnormalities, and in the absence of treatment, disease progression results in seizures, stupor, and coma. Retinochoroiditis, pneumonia, and evidence of other multifocal organ system involvement can be seen after dissemination of infection but are rare manifestations in this patient population. CT scan or MRI of the brain will typically show multiple contrast-enhancing lesions, often with associated edema (106,107,114--116). Positron emission tomography (PET) (115) or single-photon emission computed tomography (SPECT) scanning (116) might be helpful for distinguishing between TE and primary central nervous system (CNS) lymphoma, but no imaging technique is completely specific. Diagnosis HIV-1--infected patients with TE are almost uniformly seropositive for anti-toxoplasma IgG antibodies (106--108, 117). The absence of IgG antibody makes a diagnosis of toxoplasmosis unlikely but not impossible. Anti-toxoplasma IgM antibodies are usually absent. Quantitative antibody titers are not diagnostically useful. Definitive diagnosis of TE requires a compatible clinical syndrome; identification of one or more mass lesions by CT, MRI, or other radiographic testing; and detection of the organism in a clinical sample. For TE, this requires a brain biopsy, which is most commonly performed by a stereotactic CT-guided needle biopsy. Organisms are demonstrable with hematoxylin and eosin stains, though immunoperoxidase staining by experienced laboratories might increase sensitivity (118). Detection of T. gondii by polymerase chain reaction (PCR) in cerebrospinal fluid has produced disappointing results; although specificity is high (96%--100%), sensitivity is low (50%) and the results usually are negative once specific anti-toxoplasma therapy has been started (119,120). In the presence of neurologic disease, the differential diagnosis (121) includes CNS lymphoma, mycobacterial infection (especially TB), fungal infection (e.g. cryptococcosis), Chagas disease, bacterial abscess, and rarely PML, which can be distinguished on the basis of imaging studies (PML lesions typically involve white matter rather than gray matter, are noncontrast enhancing, and indicate no mass effect). Certain clinicians rely initially on an empiric diagnosis, which can be established as an objective response, on the basis of clinical and radiographic improvement, to specific anti-T. gondii therapy in the absence of a likely alternative diagnosis. Brain biopsy is reserved for patients failing to respond to specific therapy. Treatment Recommendations The initial therapy of choice consists of the combination of pyrimethamine plus sulfadiazine plus leucovorin (122--125) (AI). Pyrimethamine penetrates the brain parenchyma efficiently even in the absence of inflammation (126). Use of leucovorin prevents the hematologic toxicities associated with pyrimethamine therapy (127,128). The preferred alternative regimen for patients unable to tolerate or who fail to respond to first-line therapy is pyrimethamine plus clindamycin plus leucovorin (122,123) (AI). TMP-SMX was reported in a small (77 patient) randomized trial to be effective and better tolerated than pyrimethamine-sulfadiazine (129). On the basis of less in vitro activity and less experience with this regimen, pyrimethamine plus sulfadiazine with leucovorin is the preferred therapy (BI). For patients who cannot take an oral regimen, no well-studied options exist. No parenteral formulation of pyrimethamine exists; the only widely available parenteral sulfonamide is the sulfamethoxazole component of TMP-SMX. Therefore, certain specialists will treat severely ill patients requiring parenteral therapy initially with oral pyrimethamine plus parenteral TMP-SMX or parenteral clindamycin (CIII). At least three regimens have activity in the treatment of TE in at least two, nonrandomized, uncontrolled trials, although their relative efficacy compared with the previous regimens is unknown: 1) atovaquone (with meals or oral nutritional supplements) plus pyrimethamine plus leucovorin (130) (BII); 2) atovaquone combined with sulfadiazine or, for patients intolerant of both pyrimethamine and sulfadiazine, as a single agent (130) (BII) (if atovaquone is used alone, measuring plasma levels might be helpful given the high variability of absorption of the drug among different patients; plasma levels of >18.5 µg/mL are associated with an improved response rate) (131--133); and 3) azithromycin plus pyrimethamine plus leucovorin daily (134,135) (BII). The following regimens have been reported to have activity in the treatment of TE in small cohorts of patients or in case reports of one or a few patients: clarithromycin plus pyrimethamine (136) (CIII); 5-fluoro-uracil plus clindamycin (137) (CIII), dapsone plus pyrimethamine plus leucovorin (138) (CIII); and minocycline or doxycycline combined with either pyrimethamine plus leucovorin, sulfadiazine, or clarithromycin (139,140) (CIII). Although the clarithromycin dose used in the only published study was 1 g twice a day, doses >500 mg have been associated with increased mortality in HIV-1--infected patients treated for disseminated MAC. Doses >500 mg twice a day should not be used (DIII). Acute therapy should be continued for at least 6 weeks, if there is clinical and radiologic improvement (106--109) (BII). Longer courses might be appropriate if clinical or radiologic disease is extensive or response is incomplete at 6 weeks. Adjunctive corticosteroids (e.g. dexamethasone) should be administered when clinically indicated only for treatment of a mass effect associated with focal lesions or associated edema (BIII). Because of the potential immunosuppressive effects of corticosteroids, they should be discontinued as soon as clinically feasible. Patients receiving corticosteroids should be closely monitored for the development of other OIs, including cytomegalovirus retinitis and TB disease. Anticonvulsants should be administered to patients with a history of seizures (AIII), but should not be administered prophylactically to all patients (DIII). Anticonvulsants, if administered, should be continued at least through the period of acute therapy. Monitoring and Adverse Events Changes in antibody titers are not useful for monitoring responses to therapy. Patients should be routinely monitored for adverse events and clinical and radiologic improvement (AIII). Common pyrimethamine toxicities include rash, nausea, and bone-marrow suppression (neutropenia, anemia, and thrombocytopenia) that can often be reversed by increasing the dose of leucovorin to 50--100 mg/day administered in divided doses (CIII). Common sulfadiazine toxicities include rash, fever, leukopenia, hepatitis, nausea, vomiting, diarrhea, and crystalluria. Common clindamycin toxicities include fever, rash, nausea, diarrhea (including pseudomembranous colitis or diarrhea related to Clostridium difficile toxin), and hepatotoxicity. Common TMP-SMX toxicities include rash, fever, leukopenia, thrombocytopenia, and hepatotoxicity. Drug interactions between anticonvulsants and antiretroviral agents should be carefully evaluated and doses adjusted according to established guidelines. Management of Treatment Failure A brain biopsy, if not previously performed, should be strongly considered for patients who fail to respond to initial therapy (BII) as defined by clinical or radiologic deterioration during the first week despite adequate therapy or lack of clinical improvement within 2 weeks. For those who undergo brain biopsy and have confirmed histopathologic evidence of TE, a switch to an alternative regimen as previously described should be considered (BIII). Recurrence of disease during secondary maintenance therapy following an initial clinical and radiographic response is unusual if patients adhere to their regimen. Prevention of Recurrence Patients who have successfully completed a 6-week course of initial therapy for TE should be administered lifelong secondary prophylaxis (i.e., chronic maintenance therapy) (141--143) unless immune reconstitution occurs because of ART (AI). Adult and adolescent patients appear to be at low risk for recurrence of TE when they have successfully completed initial therapy for TE, remain asymptomatic with respect to signs and symptoms of TE, and have a sustained (i.e., >6 months) increase in their CD4+ T lymphocyte counts to >200 cells/µL on ART (144,145). The numbers of such patients who have been evaluated remain limited. On the basis of these observations and inference from more extensive data about safety of discontinuing secondary prophylaxis for other OIs during advanced HIV-1 disease, discontinuing chronic maintenance therapy among such patients is a reasonable consideration (CIII). Certain health-care providers would obtain an MRI of the brain as part of their evaluation to determine whether discontinuation of therapy is appropriate and might be reluctant to stop therapy if any mass lesion or contrast enhancement persists (CIII). Secondary prophylaxis should be started again if the CD4+ T lymphocyte count decreases to <200 cells/µL (AIII). Special Considerations During Pregnancy Documentation of maternal T. gondii serologic status should be obtained during pregnancy. Indications for treatment of T. gondii during pregnancy should be based on confirmed or suspected symptomatic disease in the mother. Pediatric care providers should be informed if the HIV-1--infected mother is seropositive for T. gondii infection to allow evaluation of the neonate for evidence of congenital infection. Pregnant HIV-1--infected women with suspected or confirmed primary T. gondii infection during pregnancy should be managed in consultation with a maternal-fetal medicine or other appropriate specialist (146) (BIII). Treatment should be the same as in nonpregnant adults (BIII). Although pyrimethamine has been associated with birth defects in animals, limited human data have not suggested an increased risk for defects and, therefore, it can be administered to pregnant women (147,148). Pediatric providers should be notified if sulfadiazine is continued until delivery because its use might increase the risk for neonatal hyperbilirubinemia and kernicterus (148). Although perinatal transmission of T. gondii normally occurs only with acute infection in the immunocompetent host, case reports have documented occurrences of transmission with reactivation of chronic infection in HIV-1--infected women with severe immunosuppression (147,149). Because the risk for transmission with chronic infection appears low, routine evaluation of the fetus for infection with amniocentesis or cordocentesis is not indicated. Detailed ultrasound examination of the fetus specifically evaluating for hydrocephalus, cerebral calcifications, and growth restriction should be done for HIV-1--infected women with suspected primary or symptomatic reactivation of T. gondii during pregnancy. CryptosporidiosisEpidemiology Cryptosporidiosis is caused by Cryptosporidium species, a group of protozoan parasites that infect the small bowel mucosa, and in immunosuppressed persons, the large bowel and extraintestinal sites. Those at greatest risk for disease are patients with advanced immunosuppression (i.e., CD4+ T lymphocyte counts generally <100 cells/µL) (150). The three most common species infecting humans are C. hominis (formerly C. parvum genotype 1 or human genotype), C. parvum (formerly C. parvum genotype 2 or bovine genotype), and C. meleagridis. In addition, infections with C. canis, C. felis, C. muris, and Cryptosporidium pig genotype have been reported in immunocompromised patients. Preliminary analyses indicate that some zoonotic species might have a stronger association with chronic diarrhea than C. hominis. However, whether the different Cryptosporidium species are associated with differences in severity of disease or response to therapy is unknown. In developed countries with low rates of environmental contamination where potent ART is widely available, cryptosporidiosis occurs at an incidence rate of <1 per 100 person-years among persons with AIDS. Transmission occurs through ingestion of Cryptosporidium oocysts. C. hominis infects only humans, and C. parvum infects humans and other large mammals (e.g., cows and sheep). C. meleagridis infects avians (e.g., turkeys and chickens) and humans. Feces from infected animals, including humans, can contaminate water supplies and recreational water with viable oocysts despite standard chlorination (90). Person-to-person transmission, primarily among men who engage in oral-anal sex, also has been observed. Young children with cryptosporidial diarrhea also might infect adults, especially during diapering. Scrupulous handwashing, use of barriers during anal sex, and other hygiene measures might help prevent person-to-person transmission. Clinical Manifestations The most common presentation of cryptosporidiosis is the acute or subacute onset of profuse, nonbloody watery diarrhea, frequently accompanied by nausea, vomiting, and lower abdominal cramping (151). Fever is present in approximately one third of patients. Malabsorption is often present. The epithelium of both the biliary tract and the pancreatic duct can be infected with Cryptosporidium. Cholangitis and pancreatitis occur among patients with prolonged disease (152). Diagnosis Cryptosporidium species cannot be cultivated in vitro. Diagnosis of cryptosporidiosis is primarily based on microscopic identification of the oocysts in stool or tissue. Oocysts stain red with varying intensities with a modified acid-fast technique; this technique allows for differentiation of the Cryptosporidium oocysts from yeasts that are similar in size and shape but are not acid fast. Oocysts also can be detected by direct immunofluorescent or enzyme-linked immunosorbent assays (153). No consensus exists on the optimal oocyst detection method in fecal samples. The modified acid-fast stain and a fluorescein labeled monoclonal antibody technique indicate comparability for diarrheal samples, but the immunofluorescent method is probably preferable for formed stool specimens. Cryptosporidium species and genotype identification requires molecular methods (e.g., PCR followed by sequencing). Cryptosporidial enteritis can be diagnosed on small intestinal biopsy sections by identification of developmental stages of Cryptosporidium organisms, found individually or in clusters, on the brush border of the mucosal epithelial surfaces. Organisms project into the lumen because of their intracellar but extracytoplasmic characteristics and appear basophilic with hematoxylin and eosin staining. Electron microscopy allows resolution of cellular detail. Among persons with profuse diarrheal illness, a single stool specimen is usually adequate for diagnosis. Among persons with less severe disease, repeat stool sampling is recommended, although no controlled studies have demonstrated the utility of three consecutive stool samples as is the case in Giardia duodenalis infection. Treatment Recommendations ART with immune restoration (an increase of CD4+ T lymphocyte count to >100 cells/µL) is associated with complete resolution of cryptosporidiosis (154,155), and all patients with cryptosporidiosis should be offered ART as part of the initial management of their infection (AII). No consistently effective pharmacologic or immunologic therapy directed specifically against C. parvum exists. Approximately 95 interventional agents have been tried for the treatment of cryptosporidiosis with no consistent success. Paromomycin, a nonabsorbable aminoglycoside that is indicated for the treatment of intestinal amebiasis, is effective in high doses for the treatment of cryptosporidiosis in animal models (156). A meta-analysis of 11 published paromomycin studies in humans reported a response rate of 67%. However, relapse was common in certain studies, with long-term success rates of only 33%. Two randomized controlled trials have compared paromomycin with placebo among patients with AIDS and cryptosporidiosis; modest, but statistically significant improvement in symptoms and oocyst shedding was demonstrated in one, but no difference from placebo was observed in the other (157,158). A small open-label study suggested a substantial benefit of paromomycin when used in combination with azithromycin, but few cures were noted (159). Therefore, efficacy data do not support a recommendation for the use of paromomycin for therapy, although the drug appears to be safe (CIII). Nitazoxanide, an orally administered nitrothiazole benzamide, has in vivo activity against a broad range of helminths, bacteria, and protozoa, including cryptosporidia (160--162). A short-term study among patients with HIV-1 infection documented increased cure rates compared with controls (based on clearance of organisms from stool and reduced rates of diarrhea) among patients with CD4+ T lymphocyte counts >50 cells/µL, but not in those with CD4+ T lymphocyte counts <50 cells/µL (161). Available data do not warrant a definite recommendation for use of this agent in this setting, but the drug has been approved by the U.S. Food and Drug Administration (FDA) for use in children and is expected to be approved for use in adults (CIII). Treatment of persons with cryptosporidiosis should include symptomatic treatment of diarrhea (AIII). Rehydration and repletion of electrolyte losses by either the oral or intravenous route is important. Severe diarrhea, which might be >10 L/day among patients with AIDS, often requires intensive support. Aggressive efforts at oral rehydration should be made with oral rehydration solutions that contain glucose, sodium bicarbonate, potassium, magnesium, and phosphorus (AIII). Treatment with antimotility agents can play an important adjunctive role in therapy, but these agents are not consistently effective (BIII). Loperamide or tincture of opium will often palliate symptoms. Octreotide, a synthetic octapeptide analog of naturally occurring somatostatin that is approved for the treatment of secreting tumor induced diarrhea, is no more effective than other oral antidiarrheal agents, and is generally not recommended (162) (DII). Monitoring and Adverse Events Patients should be closely monitored for signs and symptoms of volume depletion, electrolyte and weight loss, and malnutrition and should receive supportive treatment. Total parenteral nutrition might be indicated in certain patients (CIII). Management of Treatment Failure Supportive treatment and optimizing ART to achieve full virologic suppression are the only feasible approaches to the management of treatment failure (CIII). Prevention of Recurrence No drug regimens are proven to be effective in preventing the recurrence of cryptosporidiosis. Special Considerations During Pregnancy As with nonpregnant woman, initial treatment efforts should rely on rehydration and initiation of ART. Pregnancy should not preclude the use of ART. MicrosporidiosisEpidemiology Microsporidia organisms are protists related to fungi, defined by the presence of a unique invasive organelle consisting of a single polar tube that coils around the interior of the spore (163,164). They are ubiquitous organisms and are likely zoonotic and/or waterborne in origin (165). The microsporidia reported as pathogens in humans include Encephalitozoon cuniculi, Encephalitozoon hellem, Encephalitozoon (Septata) intestinalis, Enterocytozoon bieneusi, Trachipleistophora hominis, Trachipleistophora anthropopthera, Pleistophora species, P. ronneeafyi, Vittaforma (Nosema) corneae, Microsporidium sp., Nosema ocularum, Brachiola (Nosema) connori, Brachiola vesiculatum, and Brachiola (Nosema) algerae (163--169). In the pre-ART era, reported prevalence rates of microsporidiosis varied between 2% and 70% among HIV-1--infected patients with diarrhea, depending on the diagnostic techniques employed and the patient population described (163--166). The incidence of microsporidiosis has declined dramatically with the widespread use of effective ART. In the immunosuppressed host, microsporidiosis is most commonly observed when the CD4+ T lymphocyte count is <100 cells/µL (163--166). Clinical Manifestations The most common manifestation of microsporidiosis is gastrointestinal tract infection with diarrhea; however, encephalitis, ocular infection, sinusitis, myositis, and disseminated infection are also described (163--166). Clinical syndromes might vary by infecting species. Enterocytozoon bieneusi is associated with malabsorption, diarrhea, and cholangitis. Encephalitozoon cuniculi is associated with hepatitis, encephalitis, and disseminated disease. Encephalitozoon (Septata) intestinalis is associated with diarrhea, disseminated infection, and superficial keratoconjuctivitis. Encephalitozoon hellem is associated with superficial keratoconjunctivitis, sinusitis, respiratory disease, prostatic abscesses, and disseminated infection. Nosema, Vittaforma, and Microsporidium are associated with stromal keratitis following trauma in immunocompetent hosts. Pleistophora, Brachiola, and Trachipleistophora are associated with myositis. Trachipleistophora is associated with encephalitis and disseminated disease. Diagnosis Although microsporidia belonging to the genera Encephalitozoon, Brachiola (B. algerae), Vittaforma (V. corneae), and Trachipleistophora have been cultivated in vitro, E. bieneusi has not been successfully cultivated in vitro. Effective morphologic demonstration of microsporidia by light microscopy can be accomplished by staining methods that produce differential contrast between the spores of the microsporidia and the cells and debris in clinical samples (e.g., stool). In addition, because of the small size of the spores (1--5 mm), adequate magnification (e.g., 1,000X) is required for visualization. Chromotrope 2R, calcofluor white (fluorescent brightener), and Uvitex 2B (fluorescent brightener) are useful as selective stains for microsporidia in stool and other body fluids (167--169). In biopsy specimens, microsporidia can be visualized with Giemsa, Brown-Hopps Gram stain, acid-fast staining, Warthin-Starry silver staining, hematoxylin and eosin, or Chromotrope 2A (169). In gastrointestinal disease, examination of three stools with chromotrope and chemofluorescent stains is often sufficient for diagnosis. If stool examination is negative and microsporidiosis is suspected, a small bowel biopsy should be performed. If the etiologic agent is encephalitozoonidae or Trachipleistophora, examination of urine often reveals the organism. Determination of the species of microsporidia causing disease can be made by the morphology of the organism demonstrated by transmission electron microscopy or by PCR using species or genus specific primers (169). Treatment Recommendations ART with immune restoration (an increase of CD4+ T lymphocyte count to >100 cells/µL) is associated with resolution of symptoms of enteric microsporidiosis, including that caused by E. bieneusi (170--172). All patients should be offered ART as part of the initial management of their infection (AII). Nevertheless, data indicate that microsporidia are suppressed but not eliminated (171). No specific therapeutic agent is active against E. bieneusi infection. A controlled clinical trial suggests that E. bieneusi might respond to oral fumagillin (60 mg/day), a water insoluble antibiotic made by Aspergillus fumigatus (173,174) (BII). However, fumagillin is not available for systemic use in the United States (173,174). One report indicates that 60 days of nitazoxanide might resolve chronic diarrhea caused by E. bieneusi in the absence of ART (175). However, the effect might be minimal among patients with low CD4+ T cell counts. Nitazoxanide is approved for use among children and is expected to be approved by the FDA for use among adults. Albendazole and fumagillin have demonstrated consistent activity against other microsporidia in vitro and in vivo (176--181). Albendazole, a benzimidazole that binds to b-tubulin, has activity against many species of microsporidia, but it is not effective for Enterocytozoon infections, although fumagillin has activity in vitro and in vivo. Albendazole is recommended for initial therapy of intestinal and disseminated (not ocular) microsporidiosis caused by microsporidia other than E. bieneusi (178,181) (AII). Itraconazole also might be useful in disseminated disease when combined with albendazole especially in infections caused by Trachipleistophora or Brachiola (CIII). Ocular infections caused by microsporidia should be treated with topical Fumidil B (fumagillin bicylohexylammonium) in saline (to achieve a concentration of 70 mg/mL of fumagillin) (180) (BII). Topical fumagillin is the only formulation available for treatment in the United States and is investigational. Although clearance of microsporidia from the eye can be demonstrated, the organism often is still present systemically and can be detected in the urine or in nasal smears. In such cases, the use of albendazole as a companion systemic agent is recommended (BIII). Metronidazole and atovaquone are not active in vitro or in animal models and should not be used to treat microsporidiosis (DII). Fluid support should be offered if diarrhea has resulted in dehydration (AIII). Malnutrition and wasting should be treated with nutritional supplementation (AIII). Monitoring and Adverse Events Albendazole side effects are rare but hypersensitivity (rash, pruritis, fever), neutropenia (reversible), CNS effects (dizziness, headache), gastrointestinal disturbances (abdominal pain, diarrhea, nausea, vomiting), hair loss (reversible), and elevated hepatic enzymes (reversible) have been reported. Albendazole is not carcinogenic or mutagenic. Topical fumagillin has not been associated with substantial side effects. Oral fumagillin has been associated with thrombocytopenia, which is reversible on stopping the drug. Management of Treatment Failure Supportive treatment and optimizing ART to attempt to achieve full virologic suppression are the only feasible approaches to the management of treatment failure (CIII). Prevention of Recurrence Treatment for ocular microsporidiosis should be continued indefinitely because recurrence or relapse might follow treatment discontinuation (BIII). Whether treatment can be safely discontinued after immune restoration with ART is unknown, although it is reasonable, on the basis of the experience with discontinuation of secondary prophylaxis (chronic maintenance therapy) for other opportunistic infections during advanced HIV-1 disease, to discontinue chronic maintenance therapy if patients remain asymptomatic with regard to signs and symptoms of microsporidiosis and have a sustained (e.g. >6 months) increase in their CD4+ T lymphocyte counts to levels >200 cells/µL after ART (CIII). Special Considerations During Pregnancy Among animals (i.e., rats and rabbits), albendazole is embryotoxic and teratogenic at dosages of 30 mg/kg body weight. Therefore, albendazole is not recommended for use among pregnant women (DIII). However, well-controlled studies in human pregnancy have not been performed. Systemic fumagillin has been associated with increased resorption and growth retardation in rats. No data on use in human pregnancy are available. However, because of the antiangiogenic effect of fumagillin, this drug should not be used among pregnant women (EIII). Topical fumagillin has not been associated with embryotoxic or teratogenic effects among pregnant women and might be considered when therapy with this agent is appropriate (CIII). Mycobacterium tuberculosis DiseaseEpidemiology In the United States, overall case rates of TB disease are declining with approximately 15,000 new cases reported in 2002 (182). HIV testing is recommended for suspected or confirmed cases of TB, but this is not uniformly practiced. Therefore, the percentage of TB patients with HIV-1 infection in the United States can only be estimated. In 1999, approximately 10% of all TB cases in the United States were known to be HIV-1 infected. The World Health Organization (WHO) estimates that TB is the cause of death for 11% of all AIDS patients (183). The percentage and absolute number of patients with TB disease who are HIV-1 infected is declining in the United States because of improved infection-control practices and better diagnosis and treatment of both HIV-1 infection and TB. With increased voluntary counseling and testing and the increasing use of treatment for latent TB infection, TB disease will probably continue to decrease among HIV-1--infected persons in the United States and Western Europe (184). Persons at high risk for TB in the United States include injection-drug users, persons from high prevalence countries, and those who live or work in congregate settings. TB disease occurs among HIV-1--infected persons at all CD4+ T lymphocyte counts. The clinical manifestations might be altered depending on the degree of immunosuppression. Those with more advanced immunosuppression (CD4+ T lymphocyte count <200 cells/µL) are more likely to have extrapulmonary or disseminated disease. In areas where TB is endemic, certain patients have higher CD4+ T lymphocyte counts at the time HIV-1--related TB disease develops; in countries with low rates of TB disease (e.g., United States and countries in Western Europe), more patients have advanced HIV-1 disease at the time TB develops. TB disease in persons with HIV-1 infection can develop immediately after exposure (i.e., primary disease) or as a result of progression after establishment of latent TB infection (i.e., reactivation disease). Primary TB has been reported in certain group outbreaks, particularly in persons with advanced immune suppression, and might account for one third or more of cases of TB disease in the HIV-infected population (185). Progression to disease among those with latent TB infection is more likely among HIV-1--infected than in HIV-uninfected persons (186). HIV-uninfected persons with a positive tuberculin skin test (TST) result have a 5%--10% lifetime risk for developing TB, compared with a 7%--10% annual risk in the HIV-1--infected person with a positive TST result. Patients with TB disease have higher HIV-1 viral loads and a more rapid progression of their HIV-1 illness than comparable HIV-1--infected patients without TB (187). Clinical Manifestations The clinical, radiographic, and histopathologic presentation of HIV-1--related TB disease is heavily influenced by the degree of immunodeficiency (188,189). With CD4+ T lymphocyte counts >350 cells/µL, HIV-1--related TB appears like TB among HIV-uninfected persons. The majority of patients have disease limited to the lungs, and common chest radiographic manifestations include upper lobe fibronodular infiltrates with or without cavitation (190). However, extrapulmonary disease is more common in HIV-1--infected persons than in non-HIV--infected persons. When extrapulmonary disease occurs in HIV-1--infected persons, clinical manifestations are not substantially different from those described in HIV-uninfected patients. With increasing degrees of immunodeficiency, extrapulmonary TB, with or without pulmonary involvement, becomes more common. At CD4+ T lymphocyte counts <50 cells/µL, extrapulmonary TB (pleuritis, pericarditis, and meningitis) is common. Among severely immunocompromised patients, TB can be a severe systemic disease with high fevers, rapid progression, and sepsis syndrome. The chest radiographic findings of TB disease in advanced AIDS are markedly different compared with those among patients with less severe HIV-1 infection; lower lobe, middle lobe, and miliary infiltrates are common and cavitation is less common. Patients with HIV-1 infection and pulmonary TB can have sputum smears and culture results positive for acid-fast bacilli (AFB) or M. tuberculosis, respectively, even with a normal chest radiograph. Histopathological findings are also affected by the degree of immunodeficiency. Patients with relatively intact immune function have typical granulomatous inflammation associated with TB disease. With progressive immunodeficiency, granulomas become poorly formed or can be completely absent (189). Diagnosis Suspicion of TB, and assiduous efforts to obtain appropriate diagnostic specimens are important in diagnosing HIV-1--related TB disease. The evaluation of suspected HIV-1-related TB should always include a chest radiograph. Sputum samples for AFB smear and culture should be obtained from patients with pulmonary symptoms, cervical adenopathy, or chest radiographic abnormalities. Sputum samples from a substantial fraction of cases of pulmonary TB are negative by direct smear microscopy. Nucleic-acid amplification (NAA) tests are useful in providing rapid identification of M. tuberculosis from sputum smear-positive specimens, but false-negative results can occur among patients with TB disease. The positive predictive value of NAA tests are decreased in persons who have sputum smear-negative results. Among patients with signs of extrapulmonary TB, needle aspiration of skin lesions, nodes, pleural, or pericardial fluid might allow for rapid diagnosis, culture, and susceptibility testing. Tissue biopsy is helpful among patients with negative fine-needle aspirates. Among patients with signs of disseminated disease, mycobacterial blood culture might allow a definitive diagnosis. Mycobacterial blood culture is more sensitive for diagnosis of TB among severely immunodeficient patients. Among patients with relatively intact immune function, the yield of sputum smear and culture examinations is similar to that of HIV-uninfected adults, with positive smear results being more common among patients with cavitary pulmonary involvement (191). TST is positive in the majority of patients with pulmonary disease and CD4+ T lymphocyte counts >200 cells/µL. Among patients with more severe immunodeficiency, sputum smear and culture examinations become somewhat less sensitive, and TST has limited diagnostic value because it is often negative (192). However, the yield of mycobacterial stain and culture of specimens from extrapulmonary sites (node aspirates and pleural and pericardial fluid) is higher among patients with advanced immunodeficiency compared with HIV-uninfected adults (193--195). Smear-positive specimens from these sites probably represent a high burden of organisms resulting from lack of effective immune response to mycobacteria and inability to limit mycobacterial replication and kill the organisms. A positive smear result in any of these specimens (sputum, needle aspirate, tissue biopsy) represents some form of mycobacterial disease but does not always represent TB. However, because TB is the most virulent mycobacterial pathogen and can be spread from person to person if pulmonary involvement is present, patients with smear-positive results should be treated for TB disease until definitive mycobacterial species identification is made. Drug susceptibility testing and adjustment of the treatment regimen based on the results are critical to the successful treatment of TB and to prevention of transmission of drug-resistant M. tuberculosis in the community. Therefore, for all patients with TB disease, testing for susceptibility to first line agents (isoniazid [INH], rifampin [RIF], and ethambutol [EMB]) should be performed, regardless of the source of the specimen. Pyrazinamide (PZA) susceptibility testing should be performed on an initial isolate if there is a sufficiently high prevalence of PZA resistance in the community. Second-line drug susceptibility testing should be performed only in reference laboratories and should be limited to specimens from patients who have had previous therapy, who are contacts of patients with drug-resistant TB disease, who have demonstrated resistance to rifampin or to other first-line drugs, or who have positive cultures after >3 months of treatment (185). Treatment Recommendations Treatment of HIV--1-related TB disease should follow the general principles developed for TB treatment among non-HIV--infected persons (AI). Early diagnosis and treatment are critical. Because of the severity of TB disease among immunocompromised patients, directly observed therapy (DOT) is strongly recommended for patients with HIV-1--related TB (AI). Multiple drugs and DOT are used to provide effective therapy, to prevent acquired drug resistance during treatment, and to allow cure with a relatively short course of treatment (6--9 months). HIV-1--infected patients have other social and medical needs and treatment success is enhanced by a case-management approach, which incorporates assistance with all of these needs (enhanced DOT) in addition to providing DOT. Multiple concerns should be considered in the treatment of HIV-1--associated TB disease. First, treatment is effective, but the optimal duration of treatment is uncertain. Second, acquired drug resistance is unusual with the use of DOT, but does occur among HIV-1--infected persons. Third, the risk for acquired rifamycin resistance has led to specific recommendations about dosing frequency. Finally, the use of potent ART among patients being treated for TB is complicated by overlapping drug toxicity profiles, drug-drug interactions, and an increase in TB manifestations during immune reconstitution (paradoxical reactions). Recent studies indicate that, with careful attention to these complicating factors, the prognosis of HIV-1--associated TB disease can be substantially improved with the provision of potent ART (AII), although the optimal relative timing between anti-TB and HIV therapy is uncertain. Treatment of drug susceptible TB disease in HIV-1--infected adults should include the use of a 6-month regimen consisting of an initial phase of INH, RIF or rifabutin, PZA, and EMB given for 2 months followed by INH and RIF (or rifabutin) for 4 months when the disease is caused by organisms known or presumed to be susceptible to first-line anti-TB drugs (185) (AI). When the organism is susceptible to INH, RIF, and PZA, EMB should be discontinued (AI). The optimal duration of therapy for HIV-1--related TB disease remains controversial. Studies in developing countries have shown that patients with HIV-1--related TB respond well to standard 6-month treatment regimens, with rates of treatment failure and relapse similar to those of HIV-uninfected patients (196). However, it is unclear whether these results are applicable to patients with advanced HIV-1 disease and TB. While awaiting definitive randomized comparisons in HIV-1--infected patients with TB disease, 6 months of therapy is probably adequate for the majority of cases, but prolonged therapy (up to 9 months) is recommended (as in HIV-negative patients) for patients with a delayed clinical or bacteriological response to therapy (symptomatic or positive culture results at or after 2 months of therapy, respectively) or perhaps with cavitary disease on chest radiograph (BII). Intermittent dosing (twice- or thrice- weekly) facilitates DOT by decreasing the total number of encounters required between the patient and the provider, making observed therapy more practical to deliver. However, once- or twice-weekly dosing has been associated with an increased rate of acquired rifamycin resistance among patients with advanced HIV-1 disease (CD4+ T lymphocyte count <100 cells/µL). Acquired rifamycin resistance was relatively common with once-weekly rifapentine plus INH and also occurred in trials of twice-weekly rifabutin plus INH and twice-weekly RIF plus INH (197--199). Therefore, once-weekly rifapentine is contraindicated among HIV-1--infected patients (EI), and it is recommended that RIF- and rifabutin-based regimens be given at least three times a week for patients with TB and advanced HIV-1 disease (CD4+ T lymphocyte count <100 cells/µL) (AII). Although treatment approaches to this population need to be further evaluated in prospective trials, a prudent management strategy consists of daily DOT during the first 2 months of therapy and thrice-weekly DOT during the continuation phase of anti-TB therapy (198) (BII). Monitoring and Adverse Events Close follow-up, consisting of clinical, bacteriologic, and occasionally, laboratory and radiographic evaluations, is essential to ensure treatment success. In patients with pulmonary TB, at least one sputum specimen for microscopic examination and culture should be obtained at monthly intervals until two consecutive specimens are negative on culture (AII). Drug susceptibility tests should be repeated on isolates from patients who have positive cultures after 3 months of treatment. Patients who have positive cultures after 4 months of treatment should be considered as having failed therapy and managed accordingly. For patients with extrapulmonary TB, the frequency and types of evaluations will depend on the sites involved and the ease with which specimens can be obtained. A detailed clinical assessment should be performed at least monthly to identify possible medication intolerance and to assess adherence. As a routine, monitoring blood tests for patients being treated with first-line drugs unless baseline abnormalities were identified is unnecessary (AII). More frequent clinical and laboratory monitoring is indicated for patients with underlying liver disease, including hepatitis C co-infection. INH, RIF, and PZA all can cause drug-induced hepatitis, and the risk might be increased in patients taking other potentially hepatotoxic agents or in persons with underlying liver dysfunction. However, because of the effectiveness of these drugs (particularly INH and RIF), they should be used, if at all possible, even in the presence of preexisting liver disease (AIII). Frequent clinical and laboratory monitoring should be performed to detect any exacerbation. Independent of HIV status for all patients with TB disease, multiple treatment options exist if serum aminotransaminases are >3 times the upper limit of normal before the initiation of treatment, and the abnormalities are not thought to be caused by TB disease. One option is to use standard therapy with frequent monitoring. A second option is to treat with RIF, EMB, and PZA for 6 months, avoiding INH (BII). A third option is to treat with INH and RIF for 9 months, supplemented by EMB for the first 2 months, thereby avoiding PZA (BII). Among patients with severe liver disease, a regimen with only one hepatotoxic agent, generally RIF plus EMB, can be given for 12 months, preferably with another agent, such as a fluoroquinolone, for the first 2 months (CIII). As previously indicated, treatment might need to be lengthened for patients who are HIV-1-infected. For patients who develop worsening hepatic function on treatment, a specialist should be consulted. Tests to monitor hepatotoxicity (aminotransferases, bilirubin, and alkaline phosphatase), renal function (serum creatinine), and platelet count should be obtained for all patients started on treatment for TB. At each monthly visit, patients taking EMB should be asked about possible visual disturbances including blurred vision or scotomata. Monthly testing of visual acuity and color discrimination is recommended for patients taking doses that, on a milligram per kilogram basis, are greater than those listed in recommended doses and for patients receiving the drug for >2 months. Patients with TB disease caused by strains of M. tuberculosis resistant to at least INH and RIF (multidrug-resistant [MDR]) are at high risk for treatment failure and further acquired drug resistance. Such patients should be referred to or have consultation obtained from specialized treatment centers as identified by the local or state health departments or CDC. Although patients with strains resistant to RIF alone have a better prognosis than patients with MDR strains, they are also at increased risk for treatment failure and additional resistance and should be managed in consultation with an expert. Antiretroviral Therapy in the Management of TB Disease and Paradoxical Reactions Rifamycin drugs are essential components of short-course regimens for treatment of TB disease. However, substantial adverse pharmacologic interactions occur between rifamycins and commonly used antiretroviral drugs (e.g., PIs and NNRTIs) as a result of changes in drug metabolism resulting from induction of the hepatic cytochrome P-450 (CYP450) enzyme system (200,201). Of the available rifamycins, RIF is the most potent CYP450 inducer and rifabutin has substantially less inducing activity. Despite such interactions, a rifamycin should generally not be excluded from the TB treatment regimen among patients receiving potent ART, except in unusual circumstances (AII). Either RIF or rifabutin can be used with NRTIs (199,200). Rifabutin can be used with certain PIs or NNRTIs (other than delavirdine) and has fewer problematic drug interactions than does rifampin (Table 5). Adjustments in rifabutin or elements of the ART regimen might be necessary with certain combinations. Two antiretroviral drug regimens have been associated with a favorable outcome when administered with RIF: efavirenz (potentially using an increased dose of 800 mg/day) plus 2 NRTIs and ritonavir (600 mg twice daily) plus 2 NRTIs. Serum concentrations of nevirapine might be adequate even in the presence of concentrations of RIF associated with enzyme induction, but clinical data are lacking. RIF should not be used with nelfinavir, saquinavir, indinavir, amprenavir, atazanavir, or dual PI combinations using low dose ritonavir (<200 mg twice daily) for which dosing guidelines are not available (EII). The optimal time for initiating ART during TB treatment is unknown. Because of the risk for prolonged airborne transmission of M. tuberculosis, initiation of treatment for TB disease should never be delayed (AI). Early initiation of ART (within the first 2--4 weeks after the start of TB therapy) might decrease HIV-1 disease progression but might be associated with a relatively high incidence of side effects and paradoxical reactions (some severe enough to warrant discontinuation of both antiretroviral and anti-TB drugs). Delaying the initiation of ART for 4--8 weeks after starting antituberculous therapy has the potential advantages of being better able to ascribe a specific cause for a drug side effect, decreasing the severity of paradoxical reactions, and decreasing the adherence challenge for the patient. Until controlled studies are conducted to evaluate the optimal time for starting ART in patients with HIV-1--associated TB disease, this decision should be individualized on the basis of the patient's initial response to TB therapy, occurrence of side effects, and acceptance of multidrug ART. For these considerations, health-care providers should avoid beginning the simultaneous administration of both potent ART and combination chemotherapy for TB; most health-care providers would wait at least 4--8 weeks (BIII). Patients already receiving ART at the time treatment for TB is started require a careful assessment of the ART regimen and, if necessary, changes to ensure optimum treatment of the HIV-1 infection in the setting of TB therapy. Because of the difficulties associated with the accurate diagnosis of an adverse drug reaction and in determining the responsible agent, the first-line anti-TB drugs should not be stopped permanently without strong evidence that the anti-TB drug was the cause of the reaction. In such situations, consultation with an expert in treating TB in persons with HIV-1 infection is recommended. Patients might experience temporary exacerbation of symptoms, signs, or radiographic manifestations of TB disease after beginning anti-TB treatment. This phenomenon is termed a paradoxical (or immune reconstitution) reaction. This reaction occurs among non-HIV-1--infected persons, but it is more common among those with HIV-1 infection, particularly those treated with ART. These reactions presumably develop as a consequence of reconstitution of immune responsiveness brought about by ART or perhaps by treatment of TB itself (202--206). Signs of a paradoxical reaction can include high fevers, increase in size and inflammation of involved lymph nodes, new lymphadenopathy, expanding central nervous system lesions, worsening of pulmonary parenchymal infiltrations, and increasing pleural effusions. Such findings should be attributed to a paradoxical reaction only after a thorough evaluation has excluded other possible causes, especially TB therapy failure. A paradoxical reaction that is not severe should be treated symptomatically with nonsteroidal anti-inflammatory agents without a change in anti-TB or antiretroviral therapy (BIII). Approaches to the management of severe reactions (e.g., high fever, airway compromise from enlarging lymph nodes, enlarging serosal fluid collections, and sepsis syndrome) have not been studied. However, case reports have documented improvements with the use of prednisone or methylprednisolone used at a dose of approximately 1mg/kg body weight and gradually reduced after 1--2 weeks (202--206) (CIII). Management of Drug Resistance and Treatment Failure If resistance to INH (with or without resistance to streptomycin) is detected, INH and streptomycin, if used, should be discontinued and the patient treated with a 6-month regimen of RIF, PZA, and EMB, which is nearly as effective as the conventional INH-containing regimen (BII). Alternatively, treatment with RIF and EMB for 12 months can be used, preferably with PZA during at least the initial 2 months (BII). Treatment regimens for TB disease caused by RIF monoresistant strains are less effective, and patients infected with these strains are at increased risk for relapse and treatment failure. A minimum of 12--18 months of treatment with INH, EMB, and a fluoroquinolone (e.g., levofloxacin) with PZA administered during the first 2 months is recommended (BIII). An injectable agent (e.g., amikacin or capreomycin) might be included in the first 2--3 months for patients with severe disease. Patients with MDR-TB are at high risk for treatment failure and relapse and require especially close follow-up during (and often after) treatment. Treatment regimens for MDR-TB should be individualized, taking into account the resistance pattern, relative activities of available anti-TB agents, the extent of disease, and presence of co-morbid conditions. The management of MDR-TB is complex and should be undertaken only by an experienced specialist or in close consultation with specialized treatment centers (AIII). Prevention of Recurrence Secondary prophylaxis (chronic maintenance therapy) for patients who have successfully completed a recommended regimen of treatment for TB disease is unnecessary (DII). However, reinfection can occur. Special Considerations During Pregnancy HIV-1--infected pregnant women who do not have documentation of a negative TST result during the preceding year should be tested during pregnancy. The frequency of anergy is not increased during pregnancy, and routine anergy testing for HIV-1--infected pregnant women is not recommended (207--210). The diagnostic evaluation for TB disease in pregnant women is the same as for nonpregnant adults. Chest radiographs with abdominal shielding result in minimal fetal radiation exposure. An increase in pregnancy complications, including preterm birth, low birthweight, and intrauterine growth retardation, might be observed among pregnant women with either pulmonary or extrapulmonary TB not confined to the lymph nodes, especially when treatment is not begun until late in pregnancy (207--213). Therapy of TB disease during pregnancy should be the same as for the nonpregnant adult, but with attention given to the following considerations (BIII):
Experience during pregnancy with the majority of the second line drugs for TB is limited. MDR-TB in pregnancy should be managed in consultation with an expert. Therapy should not be withheld because of pregnancy (AIII). The following concerns should be considered when selecting second-line anti-TB drugs for use among pregnant women:
Disseminated Mycobacterium avium Complex DiseaseEpidemiology Organisms of the Mycobacterium avium complex (MAC) are ubiquitous in the environment (219--224). M. avium is the etiologic agent in >95% of patients with AIDS who develop disseminated MAC disease (219--224). An estimated 7%--12% of adults have been previously infected with MAC, although rates of disease vary in different geographic locations (220,221,224). Although certain epidemiologic associations have been identified, no environmental exposure or behavior has been consistently associated with the subsequent development of MAC disease in susceptible persons. The mode of transmission for MAC infection is thought to be through inhalation, ingestion, or inoculation through respiratory or gastrointestinal tract portals of entry. Household or close contacts of those with MAC disease do not appear to be at increased risk for experiencing disease, and person-to-person transmission is unlikely. In the absence of effective combination ART or chemoprophylaxis in those with advanced immunosuppression, the incidence of disseminated MAC disease among persons with AIDS ranges from 20%--40% (220--222). For those with a CD4+ T lymphocyte count <100 cells/µL who are receiving effective prophylaxis or those who have responded to ART with a sustained increase in CD4+ T lymphocyte count to levels >100--200 cells/µL, the overall incidence rate has been estimated at 2 cases per 100 person-years. Most cases of MAC disease occur among persons with CD4+ T lymphocyte counts <50 cells/µL. Other factors that are associated with increased susceptibility to MAC disease are high plasma HIV-1 RNA levels (>100,000 copies/mL), previous opportunistic infections (particularly CMV disease), previous colonization of the respiratory or gastrointestinal tract with MAC, and reduced in vitro lymphoproliferative immune responses to M. avium antigens, possibly reflecting defects in T-cell repertoire. Clinical Manifestations MAC disease among patients with AIDS, in the absence of ART, is generally a disseminated multiorgan infection (225--229). Early symptoms might be minimal and might precede detectable intermittent or continuous mycobacteremia by several weeks. Symptoms include fever, night sweats, weight loss, fatigue, diarrhea, and abdominal pain. Immune reconstitution inflammatory syndrome, characterized by focal lymphadenitis with fever, is a systemic inflammatory response with signs and symptoms that are clinically indistinguishable from active infection and is similar to paradoxical reactions observed with TB disease (230--232). Bacteremia is absent. The syndrome has been described among patients with subclinical or established MAC disease and advanced immunosuppression who begin ART and have a rapid and marked increase in CD4+ T lymphocyte count (>100 cells/µL). This syndrome might be benign and self-limited or might be severe and require systemic anti-inflammatory therapy to alleviate clinical symptoms. Other localized manifestations of MAC disease have been reported most commonly among persons who are receiving and who have responded to ART. Localized syndromes include cervical or mesenteric lymphadenitis, pneumonitis, pericarditis, osteomyelitis, skin or soft tissue abscesses, genital ulcers, or CNS infection. Laboratory abnormalities particularly associated with disseminated MAC disease include anemia (often out of proportion to that expected for stage of HIV-1 disease) and elevated liver alkaline phosphatase (219--225,233--235). Hepatomegaly, splenomegaly, or lymphadenopathy (paratracheal, retroperitoneal, para-aortic, or less commonly peripheral) might be identified on physical examination or by radiographic or other imaging studies. Other focal physical findings or laboratory abnormalities might occur in the context of those localized disease syndromes previously described. Diagnosis A confirmed diagnosis of disseminated MAC disease is based on compatible clinical signs and symptoms coupled with the isolation of MAC from cultures of blood, bone marrow, or other normally sterile tissue or body fluids (233--239). Use of an Isolator® (Wampole Laboratories, Cranbury, New Jersey) or a similar blood culture system and inoculation of blood into Bactec 12B liquid medium, or direct inoculation of specimens into Bactec 13A bottles (Bactec; Becton Dickinson, Sparks, Maryland), followed by radiometric detection of growth, are recommended (237). Species identification should be performed using specific DNA probes, high performance liquid chromatography, or biochemical tests. Other ancillary studies provide supportive diagnostic information, including AFB smear and culture of stool or biopsy material obtained from tissues or organs, radiographic imaging of the abdomen or mediastinum for detection of lymphadenopathy, or other studies aimed at isolation of organisms from focal infection sites. Treatment Recommendations Initial treatment of MAC disease should consist of two antimycobacterial drugs to prevent or delay the emergence of resistance (240--255) (AI). Clarithromycin is the preferred first agent (250) (AI); it has been studied more extensively than azithromycin and appears to be associated with more rapid clearance of MAC from the blood (240,250,254,255). However, azithromycin can be substituted for clarithromycin when drug interactions or clarithromycin intolerance preclude the use of clarithromcyin (AII). Ethambutol is the recommended second drug (250) (AI). Some clinicians would add rifabutin as a third drug (CI). One randomized clinical trial demonstrated that the addition of rifabutin to the combination of clarithromycin and ethambutol for the treatment of disseminated MAC disease improved survival, and in two randomized clinical trials, this approach reduced emergence of drug resistance (246,251). These studies were completed before the availability of effective ART. The addition of rifabutin should be considered in persons with advanced immunosuppression (CD4+ T lymphocyte count <50 cells/µL), high mycobacterial loads (>2 log10 colony forming units/mL of blood), or in the absence of effective ART, settings in which mortality is increased and emergence of drug resistance are most likely (CIII). If rifabutin cannot be used because of drug interactions or intolerance (Table 5), a third or fourth drug may be selected from among either the fluoroquinolones (ciprofloxacin or levofloxacin) or parenteral amikacin (Table 6), although data supporting a survival or microbiologic benefit when these agents are added have not been compelling (240--253) (CIII). Patients who have had disseminated MAC disease diagnosed and who have not previously been treated with or are not receiving potent ART should generally have ART initiated simultaneously or within 1-2 weeks of initiation of antimycobacterial therapy for MAC disease (CIII). If ART has already been instituted, it should be continued and optimized for patients with disseminated MAC disease, unless drug interactions preclude the safe concomitant use of antiretroviral and antimycobacterial drugs (CIII). Persons who have symptoms of moderate-to-severe intensity because of an immune recovery inflammatory syndrome in the setting of ART should receive treatment initially with nonsteroidal, anti-inflammatory agents (CIII). If symptoms fail to improve, short-term (4--8 weeks) systemic corticosteroid therapy, in doses equivalent to 20--40 mg of oral prednisone QD, has been successful (256,257) (CIII). Monitoring and Adverse Events Improvement in fever and a decline in quantity of mycobacteria in blood or tissue can be expected within 2--4 weeks after initiation of appropriate therapy. However, for those with more extensive disease or advanced immunosuppression, clinical response might be delayed. A repeat blood culture for MAC should be obtained 4-8 weeks after initiation of antimycobacterial therapy for patients who fail to have a clinical response to their initial treatment regimen (i.e., little or no reduction in fever or systemic symptoms). Adverse effects with clarithromycin and azithromycin include nausea, vomiting, abdominal pain, abnormal taste, and elevations of liver transaminase levels or hypersensitivity reactions. Doses of clarithromycin >1 g per day for treatment of disseminated MAC disease have been associated with increased mortality and should not be used (258) (EI). Rifabutin doses of >450 mg/day have been associated with higher risk for adverse drug interactions when used with clarithromycin or other drugs that inhibit cytochrome p450 isoenzyme 3A4 and might be associated with a higher risk for experiencing uveitis or other adverse drug reactions (259,260). Management of Treatment Failure Treatment failure is defined by the absence of a clinical response and the persistence of mycobacteremia after 4--8 weeks of treatment. Testing of MAC isolates for susceptibility to clarithromycin and azithromycin is recommended for patients who fail to microbiologically respond to initial therapy, relapse after an initial response, or develop MAC disease while receiving clarithromycin or azithromycin for prophylaxis; testing for susceptibility to clarithromycin, azithromycin, ethambutol, and rifabutin might be helpful in this setting, although the predictive value for ethambutol and rifabutin with regard to response to therapy has not been established. The majority of patients who failed clarithromycin or azithromycin primary prophylaxis in clinical trials had isolates susceptible to these drugs at the time MAC disease was detected (237,240,241,251,261). Bactec® radiometric broth macrodilution is the recommended method for testing M. avium for susceptibility to antimicrobial agents (237,250,261). Minimum inhibitory concentrations (MICs) of >32 µg/mL for clarithromycin or >256 µg/mL for azithromycin are the suggested thresholds for determination of resistance based on the Bactec® method for radiometric susceptibility testing (237,251,261). Because the number of drugs with demonstrated clinical activity against MAC is limited, results of susceptibility testing should be used to construct a new multidrug regimen consisting of at least two new drugs not previously used and to which the isolate is susceptible from among the following: ethambutol, rifabutin, ciprofloxacin or levofloxacin, or amikacin (CIII). Whether continuing clarithromycin or azithromycin in the face of resistance provides additional benefit is unknown (CIII). Clofazimine should not be used on the basis of the lack of efficacy demonstrated in randomized trials and the association with increased mortality (247,249) (EII). Other second-line agents (e.g., ethionamide, thiacetazone [not available in the United States], or cycloserine) have been anecdotally combined with these drugs as salvage regimens. However, their role in this setting is not well defined. Among patients who have failed initial treatment for MAC disease or who have antimycobacterial drug resistant MAC disease, optimizing ART is an important adjunct to second-line or salvage therapy for MAC disease (AIII). Adjunctive treatment of MAC disease with immunomodulators has not been thoroughly studied, and data are insufficient to support a recommendation for use (DIII). Interferon-gamma, tumor necrosis factor-alpha, granulocyte-macrophage colony-stimulating factor, and interleukin-12, either alone or in combination with other cytokines, appear to inhibit intracellular replication or enhance in vitro intracellular killing of M. avium (256,257,262,263). Use of these immunomodulators would be a logical adjuvant treatment for those who fail conventional antimycobacterial therapy. Prevention of Recurrence Adult and adolescent patients with disseminated MAC disease should receive lifelong secondary prophylaxis (chronic maintenance therapy) (AII), unless immune reconstitution occurs as a result of ART (250,264--268). Patients are at low risk for recurrence of MAC when they have completed a course of >12 months of treatment for MAC, remain asymptomatic with respect to MAC signs and symptoms, and have a sustained increase (e.g., >6 months) in their CD4+ T lymphocyte counts to >100 cells/µL after ART. Although the numbers of patients who have been evaluated remain limited and recurrences could occur, on the basis of these observations and on inference from more extensive data indicating the safety of discontinuing secondary prophylaxis for other opportunistic infections during advanced HIV-1 disease, discontinuing chronic maintenance therapy among such patients is reasonable (250,253,267,268) (BII). Certain health-care providers recommend obtaining a blood culture for MAC, even for asymptomatic patients, before discontinuing therapy to substantiate that disease is no longer active, but it is not clear how often a positive culture will be obtained in such patients. Secondary prophylaxis should be reintroduced if the CD4+ T lymphocyte count decreases to <100 cells/µL (AIII). Special Considerations During Pregnancy Diagnostic considerations and indications for treatment are the same as among nonpregnant adults. Azithromycin is preferred over clarithromycin as the second agent with ethambutol or rifabutin because of the occurrence of birth defects in mice and rats associated with clarithromycin (269--272) (BIII). Limited data among humans do not indicate an increased risk for defects among 122 women taking clarithromycin during the first trimester, although an increased rate of spontaneous abortions was noted (271). Limited data are available on the use of azithromycin during the first trimester in humans (271,272). Bacterial Respiratory DiseaseEpidemiology Bacterial pneumonia is a common cause of HIV-1 related morbidity (273,274). Incidence of approximately 100 cases per 1,000 HIV-1--infected persons per year have been reported, a rate much higher than in the noninfected population (273). In a study comparing rates among cohorts with similar other risk factors for bacterial pneumonia, those with HIV-1 infection were 7.8 times more likely to develop bacterial pneumonia than HIV-seronegative persons (274). For certain persons, bacterial pneumonia is a symptom of HIV-1 disease. Patients can develop serious pneumococcal infections with relatively preserved CD4+ T lymphocyte counts. The high rates of bacterial pneumonia and other pyogenic respiratory tract infections probably result from multiple factors including qualitative B-cell defects that impair the ability to produce pathogen-specific antibody, impaired neutrophil function or numbers or both, and non-HIV--related factors (e.g., cigarette smoking, use of crack cocaine, IDU, alcoholism, or liver disease). The most consistent predictor of bacterial infections is the CD4+ T lymphocyte count (275--279). The etiology of bacterial pneumonia among patients with HIV-1 infection has been reported (275--285). Consistent among these has been the relative prominence of Streptococcus pneumoniae, followed by Haemophilus influenzae, Pseudomonas aeruginosa, and Staphylococcus aureus. In the majority of studies, the pathogens of atypical pneumonia (Legionella pneumophila, Mycoplasma pneumoniae, and Chlamydia pneumoniae) are rarely encountered. On the basis of data derived from studies of pneumococcal bacteremia, infection with S. pneumoniae is 150--300 times more common among patients with HIV-1 infection than in age-matched HIV-uninfected populations (282). Recurrent pneumococcal pneumonia, either with the same or unrelated serotype, is also more common among HIV-1--infected patients, with a rate of 8%--25% within 6 months (282,283). Reinfection with a different strain is more common than relapse. In the majority of series, H. influenzae (usually nontypable) is generally the second most common cause of bacterial pneumonia (284). In patients with advanced immunosuppression, S. aureus and P. aeruginosa can cause particularly aggressive invasive pneumonias, sometimes associated with bacteremia and frequent relapses after cessation of therapy (285). As reported in pneumonia studies of non-HIV-1--infected patients, a high proportion (up to 33%) of patients with HIV-1 infection will have no specific microbiologic etiology defined. Many of these undefined cases are believed to be of possible bacterial etiology based on reviews of clinical and laboratory data, including response to antibacterial therapy. Clinical Manifestations HIV-1--infected patients with bacterial pneumonia generally present in a similar fashion to those without HIV-1 infection (i.e., acute illness characterized by chills, rigors, pleuritic chest pain, and purulent sputum). Physical findings consist of fever, tachypnea, tachycardia, rales or rhonchi, and other signs of consolidation. Lobar consolidation on chest radiograph is commonly observed and is a predictor of bacterial pneumonia, although atypical presentations with multilobar, nodular, or reticulonodular patterns are occasionally described (275--285). Patients ill over a period of weeks to months are more likely to have P. jiroveci pneumonia, TB disease, or an endemic chronic fungal infection (286). Diagnosis The pace of the respiratory disease, the underlying CD4+ T lymphocyte count, the circulating neutrophil count, and the appearance of the infiltrate should guide the diagnostic evaluation for bacterial pneumonia. At a minimum, a chest radiograph, blood cultures, a white blood cell count and, if available, a Gram's stain and culture of an adequate expectorated sputum sample, should be obtained before antibiotic administration. Because PCP is a common HIV-1--related respiratory infection and might co-exist with bacterial pneumonia, an induced sputum examination for P. jiroveci staining should be performed in the appropriate clinical settings. These would include known CD4+ T lymphocyte count <250 cells/µL, other signs of advanced immunodeficiency (e.g., thrush), a previous history of PCP or other AIDS-related condition, or diffuse infiltrates on chest radiograph. For both clinical and infection-control purposes, sputum samples (either expectorated or induced) for AFB staining and TB cultures should be obtained on all HIV-1--infected hospitalized patients with pulmonary infiltrates in the appropriate epidemiologic setting. A possible exception would be the patient who has an acute onset of an illness consistent with bacterial pneumonia, has no exposure to TB, has a previous negative TST, and who has not lived in or been exposed to high-prevalence areas for TB. In the absence of clinical improvement after initiation of antibiotic therapy and depending on the clinical history and radiographic findings, the following supplemental tests might be useful: urine antigen testing for L. pneumophila and histoplasmosis; IgM and IgG serology for M. pneumoniae and C. pneumoniae; serum cryptococcal antigen; CT scanning of the chest; and bronchoscopy with bronchoalveolar lavage and biopsy. Treatment Recommendations Therapy for HIV-1--related bacterial pneumonia should target the most commonly identified pathogens, particularly S. pneumoniae and H. influenzae. Treatment guidelines appropriate for HIV-1--uninfected patients are applicable to those with HIV-1 infection (287,288). Specific recommended regimens include either an extended spectrum cephalosporin (e.g., cefotaxime or ceftriaxone) or a fluoroquinolone with activity against S. pneumoniae (e.g., levofloxacin, moxifloxacin, or gatifloxacin) (AIII). Combination therapy with a macrolide or quinolone plus a cephalosporin should be considered for those with severe illness (AIII). For high-level penicillin-resistant isolates (MIC >4.0 mg/mL), therapy should be guided by susceptibility results. Determining whether meningitis is present is important because the recommended fluoroquinolones do not reliably attain adequate cerebrospinal fluid (CSF) levels for treating pneumococcal meningitis. Among patients with severe immunodeficiency (CD4+ T lymphocyte counts <100/mL), a known history of previous Pseudomonas infection, bronchiectasis, or relative or absolute neutropenia, broadening empiric coverage to include P. aeruginosa and other gram-negative bacilli should be considered. Possible options for therapy include ceftazidime, cefepime, piperacillin-tazobactam, a carbapenem, or high dose ciprofloxacin or levofloxacin. For ceftazidime and ciprofloxacin, other antimicrobial agents would be needed to provide optimal coverage for gram-positive infections. Monitoring and Adverse Events A clinical response (i.e., a reduction in fever and improvement in laboratory studies, physical findings, and respiratory symptoms) are generally observed 48--72 hours after initiation of appropriate therapy. Radiographic improvement might require additional time for demonstrable improvement. Management of Treatment Failure HIV-1--infected patients who fail to respond to appropriate antimicrobial therapy, as determined by a lack of reduction in fever, failure of the total WBC to return toward normal, persistent or worsening pulmonary signs, symptoms or radiographic abnormalities, progressive hypoxemia or other evidence of progressive disease, should undergo further evaluation, especially bronchoalveolar lavage or transbronchial biopsy, to search for other infectious and noninfectious causes of pulmonary dysfunction. Broader spectrum antimicrobial therapy might be required while additional diagnostic testing is pursued. Management in consultation with an infectious disease specialist is recommended. Prevention of Recurrence The strategy most effective in preventing bacterial pneumonia in HIV-1--infected patients is to optimize ART (AII). No well-documented benefit has been determined for secondary prophylaxis (chronic maintenance therapy) after successful completion of antibiotic treatment for bacterial respiratory tract infections. Adults and adolescents who have a CD4+ T lymphocyte count of >200 cells/µL should be administered a single dose of 23-valent polysaccharide pneumococcal vaccine if they have not received it during the preceding 5 years (BII). Annual administration of influenza vaccine might be useful in preventing pneumococcal superinfection of influenza respiratory tract infections (BII). Administration of antibiotic chemoprophylaxis to HIV-1--infected patients who have frequent recurrences of serious bacterial respiratory infections should be considered (CIII). TMP-SMX, administered for PCP prophylaxis and clarithromycin or azithromycin, administered for MAC prophylaxis, are appropriate for drug-sensitive organisms. However, caution is required when using antibiotics solely for preventing the recurrence of serious bacterial respiratory infections because of the potential for development of drug-resistant microorganisms and drug toxicity. Special Considerations During Pregnancy The diagnosis of bacterial respiratory tract infections among pregnant women is the same as for nonpregnant adults, with appropriate shielding of the abdomen during radiographic procedures. Bacterial respiratory tracts infections should be managed as in the nonpregnant adult, with certain exceptions. Clarithromycin should be avoided because of the occurrence of birth defects associated with its use among mice and rats (DIII). Because arthropathy has been observed among immature animals with the use of quinolones during pregnancy, quinolones are generally not recommended in pregnancy and among children aged <18 years. However, >200 cases of ciprofloxacin use in pregnancy have been reported to various pregnancy registries, and its use has not been associated with arthropathy or birth defects after in utero exposure in humans. Therefore, quinolones can be used in pregnancy for drug-resistant disease when other alternatives are not available (CIII). Pneumococcal and influenza vaccine can be administered during pregnancy, and influenza vaccine is recommended for all women who will be in the second or third trimester of pregnancy during the peak of influenza season (AIII). Because administration of vaccines might be associated with a transient rise in plasma HIV-1 RNA levels, vaccination of pregnant women is best done after ART has been initiated to minimize increases in plasma HIV-1 RNA levels that might increase the risk for perinatal HIV-1 transmission. Bacterial Enteric DiseaseEpidemiology The three most common causes of bacterial diarrhea among patients with HIV-1 infection in developed countries are Salmonella, Campylobacter, and Shigella species. Patients with HIV-1 infection are at increased risk for developing salmonellosis. Two studies in the United States and Europe reported incidence rates 20--100-fold higher than the incidence in the general population without HIV-1 infection (289--292). As with non-HIV--associated salmonellosis, the probable source for Salmonella infection is ingestion of contaminated food, in particular undercooked poultry and eggs (290). Acquisition of the infection might be facilitated by HIV-1--associated gastric achlorhydria. Campylobacter jejuni has a reported incidence among HIV-1--infected persons, particularly men who have sex with men (MSM), up to 39 times higher than in the general population (293,294). Persons with HIV-1 infection, particularly sexually active MSM, appear to be at increased risk for developing shigellosis. A population-based surveillance study conducted in 1996 found the following incidence ratios compared with the HIV-seronegative and heterosexual population: MSM and HIV-seronegative 4.9 (95% confidence interval [CI] = 2.7--8.1); heterosexual and HIV-1 infected 30.6 (95% CI = 12.8--63.0); and MSM and HIV-1 infected 35.7 (95% CI = 25.1--50.4) (295). Shigella bacteremia is more common among HIV-1--infected persons and might occur in both mild and severe cases of clinical shigellosis (296). Relapses in gastroenteritis and bacteremia after appropriate treatment have also been reported (296). Clinical Manifestations The three major clinical syndromes of salmonellosis among patients with HIV-1 infection include a self-limited gastroenteritis; a more severe and prolonged diarrheal disease, associated with fever, bloody diarrhea, and weight loss; and Salmonella septicemia, which might present with or without gastrointestinal symptoms. Bacteremia can occur with each of these syndromes and is more likely to occur among those with advanced immunosuppression (289--292). In the United States, the majority of cases of Salmonella septicemia are caused by nontyphoidal strains, in particular S. enteritidis and S. typhimurium. Because nontyphoidal Salmonella bacteremia is rare in immunocompetent hosts, its diagnosis should prompt consideration of HIV testing. An additional important feature of Salmonella bacteremia among patients with AIDS is its propensity for relapse. On the basis of data from early in the AIDS epidemic, the rate of recurrent bacteremia was approximately 45% unless chronic suppressive therapy was administered (289). Campylobacter disease among those with severe or progressive immunodeficiency is often associated with more prolonged diarrhea, invasive disease, bacteremia, and extraintestinal involvement (293,294). The development of antimicrobial resistance during therapy, often associated with clinical deterioration or relapse, is also reported more frequently among HIV-1--infected persons (297,298). Shigellosis among persons with HIV-1 infection generally causes an acute, febrile, diarrheal illness with prominent upper and lower gastrointestinal symptoms. Bloody diarrhea is more commonly observed with Shigella infection than with Salmonella infection (295,296). Diagnosis The diagnosis of bacterial enteric infection is established through cultures of stool and blood. Because of the high rate of bacteremia associated with Salmonella gastroenteritis, in particular among patients with advanced HIV-1 disease, blood cultures should be obtained from any HIV-1--infected patient with diarrhea and fever. Persons with HIV-1 are also at risk for disease caused by nonjejuni Campylobacter species, including C. fetus, C. upsaliensis, C. laridis, C. cineadi, and C. fennelliae. Although blood culture systems will generally grow these organisms, routine stool cultures performed by most laboratories will fail to identify these more fastidious Campylobacter species. Endoscopy can be diagnostically useful. If lower endoscopy is performed, ulcerations similar to those seen with cytomegalovirus colitis might be evident and can only be distinguished through histopathologic examination and culture. Treatment Recommendations Immunocompetent hosts without HIV-1 infection often do not require treatment for Salmonella gastroenteritis; the condition is self-limited and treatment might prolong the carrier state. Although no treatment trials have examined this strategy among patients with HIV-1 infection, the risk for bacteremia is sufficiently high that the majority of specialists recommend treatment of all HIV-1--associated Salmonella infections (BIII). The initial treatment of choice for Salmonella infection is a fluoroquinolone (299) (AIII). Ciprofloxacin is the preferred agent (299) (AIII); it is likely that other fluoroquinolones (levofloxacin, gatifloxacin and moxifloxacin) also would be effective in treatment of salmonellosis among HIV-1--infected persons, but these have not been well evaluated in clinical studies (BIII). The length of therapy for HIV-1--related Salmonella infection is poorly defined. For mild gastroenteritis without bacteremia, 7--14 days of treatment is reasonable in an effort to reduce the risk for extraintestinal spread (BIII). Among patients with advanced HIV-1 disease (CD4+ T lymphocyte count <200/mL) or who have Salmonella bacteremia, at least 4--6 weeks of treatment is often recommended (BIII). Depending on antibiotic susceptibility, alternatives to the fluoroquinolone antibiotics for Salmonella spp. include TMP-SMX or expanded spectrum cephalosporins (e.g., ceftriaxone or cefotaxime) (BIII). As with non-HIV--infected patients, the optimal treatment of campylobacteriosis among persons with HIV-1 infection is poorly defined. Among patients with mild disease, certain clinicians might opt to withhold therapy unless symptoms persist for more than several days. Increasing resistance to fluoroquinolones makes the choice of therapy especially problematic. For mild-to-moderate disease, initiating therapy with a fluoroquinolone (ciprofloxacin) or a macrolide (azithromycin), pending susceptibility test results, and treating for 7 days is a reasonable approach (BIII). Patients with bacteremia should be treated for at least 2 weeks (BIII), and adding a second active agent (e.g., an aminoglycoside) might be prudent (CIII). Therapy for shigellosis is indicated both to shorten the duration of illness and to prevent spread of the infection to others (299) (AIII). The recommended treatment is with a fluoroquinolone for 3--7 days (AIII). Alternatives to this treatment include TMP-SMX for 3--7 days or azithromycin for 5 days (BIII). Cases of Shigella acquired internationally have high rates of TMP-SMX resistance; in addition, HIV-1--infected persons have higher rates of adverse effects related to this agent. As a result, fluoroquinolones are preferred as first-line. Treatment of patients who have Shigella bacteremia is less well defined. Depending on the severity of infection, it might be reasonable to extend treatment to 14 days, using the agents described previously (AIII). Monitoring and Adverse Events Patients should be monitored closely for response to treatment, as defined clinically by improvement in systemic signs and symptoms and resolution of diarrhea. A follow-up stool culture to demonstrate clearance of the organism is not generally required if a complete clinical response has been demonstrated but should be considered for those who fail to clinically respond to appropriate antimicrobial therapy, or when public health considerations dictate the need to ensure microbiologic cure (e.g., health-care or food service workers). Management of Treatment Failure Treatment failure is defined by the lack of improvement in clinical signs and symptoms of diarrheal illness and the persistence of organisms in stool, blood, or other relevant body fluids or tissue after completion of appropriate antimicrobial therapy for the recommended duration. Certain patients with Salmonella bacteremia might remain febrile for 5--7 days despite effective therapy. Therefore, careful observation is required to determine the adequacy of the response. Treatment should be guided by drug susceptibility testing of isolates recovered in culture. An evaluation of other factors that might contribute to failure or relapse, such as malabsorption of oral antibiotics, a sequestered focus of infection (e.g., an undrained abscess), or adverse drug reactions that interfere with antimicrobial activity, should be undertaken as indicated. Prevention of Recurrence HIV-1--infected persons who have Salmonella bacteremia should receive long-term secondary prophylaxis (chronic maintenance therapy) to prevent recurrence. Fluoroquinolones, primarily ciprofloxacin, are usually the drugs of choice for susceptible organisms (BII). Chronic suppressive or maintenance therapy is not generally recommended for Campylobacter or Shigella infections among persons with HIV-1 infection (EIII). Household contacts of HIV-1--infected persons who have salmonellosis or shigellosis should be evaluated for persistent asymptomatic carriage of Salmonella or Shigella so that strict hygienic measures or antimicrobial therapy can be instituted and recurrent transmission to the HIV-1--infected person can be prevented (CIII). Special Considerations During Pregnancy The diagnosis of bacterial enteric infections among pregnant women is the same as among nonpregnant women. Bacterial enteric infections should be managed as in the nonpregnant adult, with several considerations. Because arthropathy has been observed among immature animals with the use of quinolones during pregnancy, quinolones are generally not recommended in pregnancy and among children aged <18 years. Therefore, expanded spectrum cephalosporins, TMP-SMX or azithromycin, depending on the organism and the results of susceptibility testing, should generally be considered as first-line therapy (CIII). However, >200 cases of ciprofloxacin use in pregnancy have been reported to various pregnancy registries, and its use has not been associated with arthropathy or birth defects after in utero exposure in humans. Therefore, quinolones can be used in pregnancy for drug-resistant disease (CIII). Neonatal-care providers should be informed of maternal sulfa therapy if used near delivery because of the theoretical increased risk to the newborn of hyperbilirubinemia and kernicterus. BartonellosisEpidemiology Bacillary angiomatosis, first recognized in 1983, and associated illnesses (e.g., peliosis hepatica) are caused by bacteria of the genus Bartonella, most commonly Bartonella henselae and Bartonella quintana (300,301). Seven other Bartonella species exist and several have been associated with bacteremia and endocarditis, but none are seen with increased frequency in HIV-1-- infected persons (300--302). Cases of bacillary angiomatosis in patients with HIV-1 infection have been linked to cat exposure. Bartonella quintana, previously known as Rochalimaea quintana, is associated with louse infestation, causes trench fever (303), and is increasingly frequent among the homeless and under conditions of poor sanitation. Bacillary angiomatosis occurs most often late in HIV-1 infection in patients with a median CD4+ T lymphocyte count of <50 cells/µL in the majority of case series (300,303). Bartonellosis is often a chronic illness with disease lasting for months to years in the majority of patients. Clinical Manifestations Bartonella species have been associated with infections involving every organ system, but the characteristic presentation is bacillary angiomatosis of the skin. Bacillary angiomatosis resembles Kaposi sarcoma. Lesions are often papular, red, with smooth or eroded surfaces, are vascular and bleed if traumatized. Nodules might be observed in the subcutaneous tissue and can erode through the skin. Bone infection has been reported, and such infections are notable in that they are lytic and painful (304). Bartonella infection of the liver produces hepatic bacillary peliosis, characterized by vascular masses in the liver or spleen. Although isolated organ systems might be the principle focus of disease, infection results from hematogenous dissemination, and systemic symptoms of fever, sweats, fatigue, malaise, weight loss, and other symptoms might accompany localized syndromes. Diagnosis Diagnosis is confirmed by histopathologic examination of tissue biopsy specimens (300--306). Lesions produce vascular proliferative histopathology; modified silver stain demonstrates numerous bacilli. Tissue Gram stain or acid-fast staining is negative. Serologic tests exist and are available through CDC (307). Serologic tests are often positive for many years before the development of symptoms, underscoring the chronicity of infection or indicating reactivation disease in the setting of immunosuppression. Bartonella spp. can be isolated from blood by using lysis centrifugation (301,303,305). The organisms are difficult to isolate from tissue. Growth requires at least 3 weeks in 5% CO2. PCR methods have been developed for the identification and speciation of Bartonella but are only available as research tools. Treatment Recommendations No randomized, controlled clinical trials have evaluated antimicrobial treatment of bartonellosis. Erythromycin and doxycycline have been used successfully to treat bacillary angiomatosis, peliosis hepatica, bacteremia, and osteomyelitis and are considered first-line treatment for bartonellosis on the basis of reported experience in case series (300--307) (AII). Therapy should last at least 3 months (AII). Doxycyline is the treatment of choice for central nervous system bartonellosis (AIII). Clarithromycin or azithromycin have been associated with clinical response in certain cases and are considered second line alternatives (BII), although treatment failures have been reported with both drugs. The beta-lactams (penicillins and first-generation cephalosporins) have no appreciable in vitro activity and are not recommended for treatment of bartonellosis (DII). Quinolones have variable in vitro activity and clinical response in case reports; as a result, they are not generally recommended as first-line therapy but might be tried as second-line alternatives (CIII). Management of Treatment Failure Among patients who fail to respond to initial treatment, one or more of the second-line alternative regimens should be considered (AIII). Among patients who relapse, lifelong therapy is recommended (AIII). Prevention of Recurrence Relapse or reinfection with Bartonella has sometimes followed a course of primary treatment. Although no firm recommendation can be made about secondary prophylaxis (chronic maintenance therapy) in this setting, long-term suppression of infection with erythromycin or doxycycline should be considered (CIII). Special Considerations During Pregnancy Pregnancy has been associated with a more severe course and possible increased risk for death with acute infection caused by B. bacilliformis in immunocompetent patients (308). No data are available on the potential impact of pregnancy on Bartonella infections among HIV-1--infected persons. Similarly, B. bacilliformis infections during pregnancy might increase the risk for spontaneous abortion and stillbirth and can be transmitted to the fetus. No data are available on the effect of other Bartonella infections on pregnancy outcome. Diagnosis of Bartonella infections in pregnant women should be the same as in nonpregnant adults. Treatment during pregnancy should be with erythromycin rather than tetracyclines because of the increased hepatotoxicity and staining of fetal teeth and bones associated with the use of tetracyclines during pregnancy (AIII). Cephalosporins are not recommended. SyphilisEpidemiology Recent reports indicate a resurgence of infections with Treponema pallidum, the etiologic agent of syphilis, among men in several U.S. cities and in Western Europe, possibly because of relaxed safer sex practices of those who view HIV-1 infection as a disease manageable if not curable with effective ART (309--314). HIV-1 infection appears to alter the diagnosis, natural history, management, and outcome of T. pallidum infection (315--318). This section focuses on specific guidelines for the management of syphilis among HIV-1--infected patients. A more comprehensive review of the recommendations for the treatment of syphilis is available (319). Clinical Manifestations The impact of HIV-1 infection on syphilis pathogenesis, disease severity, response to treatment, and long-term sequelae is not well documented. As among HIV-uninfected persons, primary syphilis commonly presents as a single painless nodule at the site of contact that rapidly ulcerates to form a classic chancre; however, among HIV-infected persons, multiple or atypical chancres occur, and primary lesions might be absent or missed. Progression to secondary syphilis generally follows 2--8 weeks after primary inoculation and reflects ongoing replication and dissemination of T. pallidum in the absence of an effective host immune response. Although more rapid progression or severe disease might be present among HIV-1--infected persons with advanced immunosuppression, the clinical manifestations are similar to those among HIV-uninfected persons. The manifestations of secondary syphilis are protean, involving virtually all organ systems. The most common manifestations appear to be macular, maculopapular, or pustular skin lesions (or condyloma lata in moist genital or intertriginous areas), usually beginning on the trunk and spreading peripherally, characteristically involving palms and soles and accompanied by generalized lymphadenopathy and constitutional symptoms of fever, malaise, anorexia, arthralgias, and headache (317--319). Secondary syphilis, particularly acute syphilitic meningitis, must be distinguished from acute primary HIV-1 infection. The previously described constitutional symptoms, along with nonfocal CNS symptoms and CSF abnormalities (e.g., lymphocytic pleocytosis with a mildly elevated CSF protein) are common to both (320--322). The signs and symptoms of secondary syphilis might persist from a few days to several weeks before resolving or evolving to latent or later stages. As among HIV-uninfected patients, latent syphilis is not associated with overt clinical signs and symptoms, but relapse of manifestations of secondary syphilis might occur, most commonly in the first 1--4 years following infection. Manifestations of "late" syphilis generally include neurosyphilis, cardiovascular syphilis, and gummatous syphilis, but might present as slowly progressive disease that can affect any organ system. Certain manifestations of neurologic complications or neurosyphilis progress more rapidly or occur earlier in the course of disease among persons with HIV-1 infection and are not truly late complications or manifestations. Asymptomatic neurosyphilis, which might be the most commonly described syndrome, is defined as the absence of symptoms but with one or more abnormalities of CSF (i.e., elevated protein, lymphocytic cellular infiltrate, or positive serologic tests). Manifestations of symptomatic neurosyphilis (i.e., meningitis or meningovascular or parenchymatous disease) among HIV-1--infected persons will probably be similar to those in the HIV-uninfected population. However, concomitant uveitis and meningitis might be more common among HIV-1--infected patients with syphilis. Diagnosis The diagnosis of syphilis depends on a variety of tests that either directly detect the organism (e.g., darkfield microscopy or direct fluorescent antibody-Treponema Pallidum (DFA-TP) or serum antibodies against it (e.g., FTA-ABS and TP-TA), or indirectly indicate the presumptive presence of T. pallidum by detecting nontreponemal antibodies generated during infection (e.g., VDRL and RPR) (317,319,323). Clinical experience indicates that concurrent HIV-1 infection probably does not change the performance of standard tests for the diagnosis of syphilis, but this concern has not been formally studied. Early-stage disease (i.e., primary, secondary and early-latent syphilis) among HIV-1--infected patients is confirmed by the identical procedures used for the HIV-uninfected populations (darkfield microscopy of a mucocutaneous lesion sample and standard serologic tests). HIV-1 infection does not decrease the sensitivity or specificity of darkfield microscopy. Responses to nontreponemal serologic tests (i.e., VDRL and RPR) might be atypical (i.e., higher, lower, or delayed) among HIV-1--infected versus HIV-uninfected patients with early-stage syphilis, but no data indicate that treponemal tests perform differently among HIV-1--infected compared with uninfected patients. Similar to HIV-uninfected persons, false-negative serologic tests have been reported among HIV-1--infected patients with documented T. pallidum infection. Therefore, if the clinical suspicion of syphilis is high and serologic tests do not confirm the diagnosis, other diagnostic procedures (e.g., biopsy, darkfield examination, or direct fluorescent antibody staining of lesion material) should be pursued. By definition, patients presenting with latent syphilis have serological evidence of disease in the absence of clinical or other laboratory abnormalities (i.e., normal CSF profiles). Patients with early-latent syphilis by definition have documented infection of <1 year; patients with late-latent syphilis have documented infection for >1 year, or the duration of infection is not known. The diagnostic testing for detection of late-stage disease (e.g., cardiovascular and gummatous syphilis) among HIV-1--infected patients is the same as for the HIV-uninfected population. Diagnosis of neurosyphilis is established by examination of the CSF, which might indicate mild mononuclear pleocytosis (10--200 cells/µL), normal or mildly elevated protein concentration, or a reactive CSF-VDRL (319,324). The CSF-VDRL is specific but not sensitive, and a reactive test establishes the diagnosis of neurosyphilis but a nonreactive test does not exclude the diagnosis. In comparison, CSF treponemal tests (e.g., the CSF FTA-ABS) are sensitive but not specific, and a nonreactive test excludes the diagnosis of neurosyphilis, but a reactive test does not establish the diagnosis. Calculated indices (e.g., ITPA-index) are of limited value in establishing the diagnosis of neurosyphilis. PCR-based diagnostic methods are not recommended as a diagnostic test for neurosyphilis. A reactive CSF-VDRL and a CSF WBC >10 cells/µL support the diagnosis of neurosyphilis; the majority of specialists would not base the diagnosis solely on elevated CSF protein concentrations in the absence of these other abnormalities. HIV-1 infection itself might be associated with mild mononuclear CSF pleocytosis (5--15 cells/µL), particularly among persons with peripheral blood CD4+ T lymphocyte counts >500 cells/µL. Establishing the diagnosis of neurosyphilis might be more difficult among such persons. If neurosyphilis cannot be excluded by a nonreactive CSF treponemal test, such persons should be treated for neurosyphilis, despite the acknowledged uncertainty of the diagnosis. Treatment Recommendations Management of HIV-1--infected patients with syphilis is similar to the management of HIV-uninfected persons with the disease (319,325,326). However, closer follow-up is recommended to detect potential treatment failures or disease progression. All patients with syphilis, regardless of disease stage, should be evaluated for clinical evidence of CNS or ocular involvement. Those with neurologic or ocular symptoms or signs should undergo CSF examination to rule out neurosyphilis. HIV-1--infected patients with late-latent syphilis, including those with syphilis of unknown duration, also should undergo CSF examination. Certain specialists recommend CSF examination for all HIV-1--infected patients with syphilis, regardless of stage. Similar to the HIV-uninfected population, HIV-1--infected patients with active tertiary syphilis (i.e., aortitis and gumma) or who fail treatment for non-neurologic syphilis should undergo CSF examination. Patients with CSF abnormalities consistent with neurosyphilis should be treated for neurosyphilis. HIV-1--infected persons with early-stage (i.e., primary, secondary, or early latent) syphilis should receive a single intramuscular (IM) injection of 2.4 million units of benzathine penicillin G (AII). Alternative therapies, including oral doxycycline, ceftriaxone, and azithromycin, have not been sufficiently evaluated in HIV-1--infected patients to warrant use as first-line treatment. If the clinical situation requires the use of an alternative to penicillin, treatment should be undertaken with close clinical monitoring (BIII). In a randomized clinical trial, amoxicillin administered with probenecid, which increases CSF amoxicillin levels, did not improve clinical outcome of early stage disease and is not recommended (325) (DII). In HIV-1--infected patients with late-latent syphilis for whom the CSF examination excludes the diagnosis of neurosyphilis, treatment with three weekly intramuscular injections of 2.4 million units benzathine penicillin G is recommended (AIII). Alternative therapy with doxycycline 100 mg by mouth twice a day for 28 days has not been sufficiently evaluated in HIV-1--infected patients to warrant use as first-line treatment. If the clinical situation requires the use of an alternative to penicillin, treatment should be undertaken with close clinical monitoring (BIII). HIV-1--infected patients with clinical evidence of late-stage (tertiary) syphilis (cardiovascular or gummatous disease) should have a CSF examination to rule out neurosyphilis before initiating therapy (AIII). The complexity of tertiary syphilis management is beyond the scope of these guidelines and providers treating tertiary disease are advised to consult an infectious disease specialist (AIII). HIV-1--infected patients with clinical or laboratory evidence of neurosyphilis (i.e., CNS involvement including otic and ocular disease, even with a normal CSF) should receive intravenous aqueous crystalline penicillin G, 18--24 million units daily, administered 3--4 million units IV every 4 hours or by continuous infusion for 10--14 days (AII) or procaine penicillin 2.4 million units IM once daily plus probenecid 500 mg orally four times a day for 10--14 days (319,327,328) (BII). HIV-1--infected patients who are allergic to sulfa-containing medications should not be administered the IM alternative because they are very likely to be allergic to probenecid (DIII). IM procaine penicillin without probenecid does not achieve sufficient penicillin levels in CSF to treat neurosyphilis. Because neurosyphilis treatment regimens are of shorter duration than those used in late-latent syphilis, certain specialists recommend following neurosyphilis treatment with 3 weeks of benzathine penicillin, 2.4 million units IM weekly. However, no consensus has been reached about the need for this practice (CIII). Among penicillin allergic patients, penicillin desensitization followed by one of the penicillin regimens listed previously is the preferred approach (BIII). However, limited data indicate that ceftriaxone (2 g daily IV for 10--14 days) might be an alternative regimen (CIII). Monitoring and Adverse Events Clinical and serologic responses to treatment of early stage (i.e., primary, secondary, and early-latent) disease should be monitored at 3, 6, 9, 12, and 24 months after therapy. Serologic responses to treatment might differ among HIV-1--infected patients compared with HIV-uninfected persons, including temporal pattern of response and proportion of subjects achieving serologically defined treatment success (at least a fourfold decrease in titer). After successful treatment for syphilis among HIV-1--infected and uninfected patients, some might remain "serofast," meaning that serum nontreponemal test titers remain reactive at low and unchanging titers, generally <1:8, for extended periods of time (up to the lifetime of the patient). The clinical significance of the serofast state is unclear, but it probably does not represent treatment failure. Serologic detection of potential re-infection should be based on at least a fourfold increase in titer above the established serofast baseline. Response to therapy of late-latent syphilis should be monitored using nontreponemal serologic tests at 3, 6, 12, 18, and 24 months to ensure at least a fourfold decline in titer. Two retrospective studies reported that concomitant HIV-1 infection was associated with poorer CSF and serologic responses to neurosyphilis therapy (326,327). Repeat CSF examination should be performed at 3 and 6 months after completion of therapy and then every 6 months until the CSF white blood cell count is normal and the CSF-VDRL is nonreactive. Because of the complex nature of neurosyphilis, treatment should be undertaken in consultation with an infectious disease specialist. Management of Treatment Failure Re-treatment of patients with early stage syphilis should be considered for those who 1) do not experience at least a fourfold decrease in serum nontreponemal test titers 6--12 months after therapy, 2) have a sustained fourfold increase in serum nontreponemal test titers after an initial reduction after treatment, or 3) have persistent or recurring clinical signs or symptoms of disease (BIII). If CSF examination does not confirm the diagnosis of neurosyphilis, such patients should receive 2.4 million units IM benzathine penicillin G administered at 1-week intervals for 3 weeks (BIII). Certain specialists have also recommended a course of aqueous penicillin G IV or procaine penicillin IM plus probenecid, as described for treatment of neurosyphilis above, in this setting (CIII). If titers fail to respond appropriately after re-treatment, repeat CSF evaluation or re-treatment might not be beneficial (CIII). Patients with late-latent syphilis should have a repeat CSF examination and be retreated if they have clinical signs or symptoms of syphilis, have a fourfold increase in serum nontreponemal test titer, or experience an inadequate serologic response (less than fourfold decline in nontreponemal test titer) within 12--24 months of therapy (BIII). If the CSF examination is consistent with CNS involvement, re-treatment should follow the neurosyphilis recommendations (AIII); those without a profile indicating CNS disease should receive a repeat course of benzathine penicillin, 2.4 million units IM weekly for 3 weeks (BIII), although certain specialists recommend following the neurosyphilis recommendations in this setting (CIII). Re-treatment of neurosyphilis should be considered if the CSF WBC count has not decreased after 6 months after completion of treatment, or if the CSF-VDRL remains reactive 2 years after treatment (BIII). Secondary Prevention and Maintenance Therapy No recommendations have been developed indicating the need for secondary prophylaxis or prolonged chronic maintenance antimicrobial therapy for syphilis in HIV-1--infected patients. Special Considerations During Pregnancy All pregnant women should be screened for syphilis at the first prenatal visit. In areas where syphilis prevalence is high or among women at high risk (e.g., uninsured, women living in poverty, commercial sex workers, and injection-drug users), testing should be repeated at 28 weeks of gestation and at delivery. All women delivering a stillborn infant after 20 weeks of gestation should also be tested for syphilis. Syphilis screening should also be offered at sites providing episodic care to pregnant women at high risk including emergency departments, jails, and prisons. No infant should leave the hospital without documentation of maternal syphilis serology status during pregnancy (329). The rate of transmission and adverse outcomes of untreated syphilis are highest with primary, secondary, and early latent syphilis during pregnancy and decrease with increasing duration of infection thereafter. Pregnancy does not appear to alter the course, manifestations, or diagnostic test results of syphilis infection among adults. The diagnosis should be made the same as among nonpregnant adults. Concurrent syphilis infection might increase the risk for perinatal transmission of HIV-1 to the infant, although an increased risk has not been consistently reported (330--333). Treatment during pregnancy should consist of the same penicillin regimen as recommended for the given disease stage among nonpregnant, HIV-1--infected adults. Because of treatment failures reported after single injections of benzathine penicillin among HIV-uninfected pregnant women (334), certain specialists recommend a second injection 1 week after the initial injection for pregnant women with early syphilis (319,335). Because of additional concerns about the efficacy of standard therapy in HIV-1--infected persons, a second injection 1 week after the first for HIV-1--infected pregnant women should be considered (BIII). No alternatives to penicillin have been proven effective and safe for treatment of syphilis during pregnancy or for prevention of fetal infection. Pregnant women who have a history of penicillin allergy should be referred for skin testing and desensitization and treatment with penicillin (319) (AIII). Erythromycin does not reliably cure fetal infection; tetracyclines should not be used during pregnancy because of hepatotoxicity and staining of fetal bones and teeth (EIII). Efficacy data with azithromycin or ceftriaxone are insufficient to support a recommendation for their use in this setting (DIII). A Jarisch-Herxheimer reaction occurring during the second half of pregnancy might precipitate preterm labor or fetal distress (336). Consideration should be given to providing fetal and contraction monitoring for 24 hours after initiation of treatment for early syphilis of pregnant women who are >20 weeks of gestation, especially in the setting of abnormal ultrasound findings indicative of fetal infection (BIII). Alternatively, women should be advised to seek obstetric attention after treatment if they notice contractions or a decrease in fetal movement. Repeat serologic titers should be performed in the third trimester and at delivery for women treated for syphilis during pregnancy. Titers can be conducted monthly for women at high risk for reinfection. The clinical and antibody response should be appropriate for the stage of disease, although the majority of women will deliver before their serologic response can be definitively assessed. Mucocutaneous CandidiasisEpidemiology Oropharyngeal and esophageal candidiasis are common (337). The majority of infection is caused by Candida albicans. Fluconazole (or azole) resistance is predominantly the consequence of previous exposure to fluconazole (or other azoles), particularly repeated and long-term exposure (338--340). In this setting, C. albicans resistance has been accompanied by a gradual emergence of non-albicans Candida species, particularly C. glabrata, as a cause of refractory mucosal candidiasis, particularly in patients with advanced immunosuppression (338,341). The occurrence of oropharyngeal or esophageal candidiasis is recognized as an indicator of immune suppression, and these are most often observed in patients with CD4+ T lymphocyte counts <200 cells/µL (337). In contrast, vulvovaginal candidiasis is common among healthy, adult women and is unrelated to HIV-1 status. Recurrent vulvovaginal candidiasis alone should not be considered a sentinel of HIV-1 infection among women. The introduction of ART has led to a dramatic decline in the prevalence of oropharyngeal candidiasis and a marked diminution in cases of refractory disease. Clinical ManifestationsOropharyngeal candidiasis is characterized by painless, creamy white, plaque-like lesions of the buccal or oropharyngeal mucosa or tongue surface. Lesions can be easily scraped off with a tongue depressor or other instrument. Less commonly, erythematous patches without white plaques can be seen on the anterior or posterior upper palate or diffusely on the tongue. Angular chelosis is also noted on occasion and may be caused by Candida. Esophageal candidiasis is occasionally asymptomatic but often presents with fever, retrosternal burning pain or discomfort, and odynophagia. Endoscopic examination reveals whitish plaques similar to those observed with oropharyngeal disease that might progress to superficial ulceration of the esophageal mucosa, with central or surface whitish exudates. Vulvovaginitis might be mild to moderate and sporadic, similar in presentation to that in normal hosts, and characterized by a creamy to yellow-white adherent vaginal discharge associated with mucosal burning and itching. In those with more advanced immunosuppression, episodes might be more severe, more frequently recurrent, of longer duration, or refractory to treatment. Diagnosis Diagnosis of oropharyngeal candidiasis is usually clinical and based on the appearance of lesions. The feature that distinguishes these from oral hairy leukoplakia is the ability to scrape off the superficial whitish plaques. If laboratory confirmation is required, a scraping for microscopic examination for yeast forms using a potassium hydroxide (KOH) preparation provides supportive diagnostic information. Cultures of clinical material identify the species of yeast present. The diagnosis of esophageal candidiasis requires endoscopic visualization of lesions with histopathologic demonstration of characteristic Candida yeast forms in tissue and culture confirmation of the presence of Candida species. The diagnosis of vulvovaginal candidiasis is based on the clinical presentation coupled with the demonstration of characteristic yeast forms in vaginal secretions examined microscopically after KOH preparation. Culture confirmation is rarely required but might provide supportive information. Because self-diagnosis of vulvovaginitis is unreliable, microscopic confirmation is required to avoid unnecessary exposure to inappropriate treatments. Treatment Recommendations Although initial episodes of oropharyngeal candidiasis can be adequately treated with topical therapy, including clotrimazole troches or nystatin suspension or pastilles (BII), oral fluconazole is as effective and, in certain studies, superior to topical therapy and is more convenient and generally better tolerated (342) (AI). Itraconazole oral solution for 7--14 days is as effective as oral fluconazole but less well tolerated (AI). Ketoconazole and itraconazole capsules are less effective than fluconazole because of their more variable absorption and should be considered second line alternatives (DII). Systemic therapy is required for effective treatment of esophageal candidiasis (AII). A 14--21-day course of either fluconazole or itraconazole solution is highly effective (AI). As with oropharyngeal candidiasis, ketoconazole and itraconazole capsules are less effective than fluconazole because of variable absorption (DII). Although caspofungin (AII) and voriconazole (AII) are effective in treating esophageal candidiasis among HIV-1--infected patients, experience is limited and fluconazole remains the preferred agent. Although symptoms of esophageal candidiasis might be mimicked by other pathogens, a diagnostic trial of antifungal therapy is often appropriate before endoscopy is undertaken to search for other causes of esophagitis. Uncomplicated vulvovaginal candidiasis is observed in 90% of HIV-1--infected women and responds readily to short-course oral or topical treatment with any of several therapies including single-dose regimens (AII):
Complicated vaginitis (prolonged or refractory episodes) is observed in approximately 10% of patients and requires antimycotic therapy for >7 days (AII). Monitoring and Adverse Events For the majority of patients, response to therapy is rapid, with improvement in signs and symptoms within 48--72 hours. Short courses of topical therapy rarely result in adverse effects, although patients might experience cutaneous hypersensitivity reactions, with rash and pruritis. Patients might experience gastrointestinal upset with oral azole treatment. Patients treated for >7--10 days with azoles might experience hepatotoxicity. If prolonged therapy is anticipated (>21 days), periodic monitoring of liver chemistry studies should be considered. Management of Treatment Failure Treatment failure is generally defined as signs and symptoms of oropharyngeal or esophageal candidiasis that persist for more than 7--14 days of appropriate therapy. Fluconazole-refractory oropharyngeal candidiasis will respond at least transiently to itraconazole solution in approximately two thirds of persons (AII). Amphotericin B oral suspension (1 mL four times daily of the 100 mg/mL suspension) is sometimes effective among patients with oropharyngeal candidiasis who do not respond to itraconazole (CIII); however, this product is not available in the United States. Intravenous amphotericin B is usually effective and can be used among patients with refractory disease (BII). Fluconazole-refractory esophageal candidiasis should be treated with caspofungin (BIII) or intravenous amphotericin B, either conventional or liposomal or lipid complex formulations (BII). Prevention of Recurrence The majority of HIV specialists do not recommend secondary prophylaxis (chronic maintenance therapy) of recurrent oropharyngeal or vulvovaginal candidiasis because of the effectiveness of therapy for acute disease, the low mortality associated with mucosal candidiasis, the potential for resistant Candida organisms to develop, the possibility of drug interactions, and the cost of prophylaxis (DIII). However, if recurrences are frequent or severe, an oral azole, fluconazole (CI), or itraconazole solution (CI) (or for recurrent vulvovaginal candidiasis, daily prophylaxis with any topical azole [CII]) should be considered. Other factors that influence choices related to such therapy include impact of recurrences on the patient's well-being and quality of life, need for prophylaxis for other fungal infections, cost, toxicities, drug interactions, nutritional status, and potential to induce drug resistance among Candida and other fungi. Prolonged use of systemically absorbed azoles, specifically among patients with low CD4+ T lymphocyte counts (i.e., <100 cells/µL) increases the risk for developing azole resistance. Adults or adolescents who have a history of one or more episodes of documented esophageal candidiasis should be considered candidates for secondary prophylaxis. Fluconazole 100--200 mg daily is appropriate (BI). However, potential azole resistance should be considered when long-term azoles are considered. Special Considerations During Pregnancy Pregnancy increases the risk for vaginal colonization with Candida species. Diagnosis of oropharyngeal, esophageal, and vulvovaginal candidiasis is the same as among nonpregnant adults. Fluconazole is teratogenic in high doses in animal studies (343,344). Among humans, four cases of an unusual cluster of defects (i.e., craniofacial and skeletal) have been reported after prolonged use at high doses in the first trimester of pregnancy (345,346). Teratogenic effects have not been described among animals at doses similar to those used in humans, and anomalies do not appear to be increased among infants born to women receiving single-dose fluconazole treatment in the first trimester (347--349). Itraconazole is teratogenic among rats and mice (i.e., skeletal defects, encephalocele, and macroglossia) at high doses (350). Similar to fluconazole, no increase in anomalies has been noted among women exposed to treatment doses in the first trimester. Invasive or refractory esophageal Candida infections should be treated the same in pregnancy as in the nonpregnant woman, with the exception that amphotericin B should be substituted for fluconazole or itraconazole (if indicated) in the first trimester if similar efficacy is to be expected (351) (BIII). CryptococcosisEpidemiology Virtually all HIV-1--associated cryptococcal infections are caused by Cryptococcus neoformans var neoformans. Before the advent of ART, approximately 5%--8% of HIV-1--infected patients in developed countries acquired disseminated cryptococcosis (352,353). The incidence has declined substantially with use of effective ART. The majority of cases of infection are observed among patients who have CD4+ T lymphocyte counts of <50 cells/µL. Clinical Manifestations Cryptococcosis among patients with AIDS most commonly occurs as a subacute meningitis or meningoencephalitis with fever, malaise, and headache (352). Classic meningeal symptoms and signs (e.g., neck stiffness or photophobia) occur in approximately one fourth to one third of patients. Certain patients might present with encephalopathic symptoms (e.g., lethargy, altered mentation, personality changes, and memory loss). Analysis of the CSF usually indicates a mildly elevated serum protein, normal or slightly low glucose, and a few lymphocytes and numerous organisms. The opening pressure in the CSF is elevated (with pressures >200 mm of water) in up to 75% of patients. Disseminated disease is a common manifestation, with or without concurrent meningitis. Approximately half of patients with disseminated disease have evidence of pulmonary rather than meningeal involvement. Symptoms and signs of pulmonary infection include cough or dyspnea and abnormal chest radiographs. Skin lesions might be observed. Diagnosis Cryptococcal antigen is almost invariably detected in the CSF at high titer in patients with meningitis or meningoencephalitis. Up to 75% of patients with HIV-1--associated cryptococcal meningitis have positive blood cultures; if disseminated or other organ disease is suspected in the absence of meningitis, a fungal blood culture is also diagnostically helpful. The serum cryptococcal antigen is also usually positive and detection of cryptococcal antigen in serum might be useful in initial diagnosis (354). Treatment Recommendations Untreated cryptococcal meningitis is fatal. The recommended initial treatment for acute disease is amphotericin B, usually combined with flucytosine, for a 2-week duration followed by fluconazole alone for an additional 8 weeks (AI). This approach is associated with a mortality of <10% and a mycologic response of approximately 70% (355,356). The addition of flucytosine to amphotericin B during acute treatment does not improve immediate outcome but is well tolerated for 2 weeks and decreases the risk for relapse (355, 356). Lipid formulations of amphotericin B appear effective. The optimal dose of lipid formulations of amphotericin B has not been determined, but AmBisome has been effective at doses of 4 mg/kg body weight/daily (356,357) (AI). After a 2-week period of successful induction therapy, consolidation therapy should be initiated with fluconazole administered for 8 weeks or until CSF cultures are sterile (355,356,358) (AI). Itraconazole is an acceptable though less effective alternative (358) (BI). Combination therapy with fluconazole (400--800 mg/daily) and flucytosine is effective for treating AIDS-associated cryptococcal meningitis (359). However, because of the toxicity of this regimen (especially myelotoxicity and gastrointestinal toxicity), it is recommended only as an alternative option for persons unable to tolerate or unresponsive to standard treatment (BII). Increased intracranial pressure might cause clinical deterioration despite a microbiologic response, probably reflects cerebral edema, and is more likely if the CSF opening pressure is >200 mm H2O (355, 360). In one large clinical trial, 93% of deaths occurring within the first 2 weeks of therapy and 40% of deaths occurring within weeks 3--10 were associated with increased intracranial pressure (360). The opening pressure should always be measured when a lumbar puncture is performed (360). The principal initial intervention for reducing symptomatic elevated intracranial pressure is repeated daily lumbar punctures (AII). CSF shunting should be considered for patients in whom daily lumbar punctures are no longer being tolerated or whose signs and symptoms of cerebral edema are not being relieved (BIII). Whether reducing opening pressure leads to a reduction in the mortality and morbidity associated with cerebral edema is unknown. No role exists for acetazolamide to reduce intracranial pressure (DIII). Monitoring and Adverse Events A repeat lumbar puncture to ensure clearance of the organism is not required for those with cryptococcal meningitis and improvement in clinical signs and symptoms after initiation of treatment. If new symptoms or clinical findings occur after 2 weeks of treatment, a repeat lumbar puncture should be performed. Serum cryptococcal antigen is not helpful in management because changes in titer do not correlate with clinical response (354). Serial measurement of CSF cryptococcal antigen might be more useful but requires repeated lumbar punctures and is not routinely recommended for monitoring response. Patients treated with amphotericin B should be monitored for dose-dependent nephrotoxicity and electrolyte disturbances. Supplemental colloidal fluids might reduce the risk for nephrotoxicity during treatment (CIII). Infusion-related adverse reactions (e.g., fever, chills, renal tubular acidosis, hypokalemia, orthostatic hypotension, tachycardia, nausea, headache, vomiting, anemia, anorexia, and phlebitis) might be ameliorated by pretreatment with acetaminophen, diphenhydramine, or corticosteroids administered approximately 30 minutes before the infusion (CIII). Lipid formulations of amphotericin B are less toxic. Azotemic patients receiving flucytosine should have their blood levels monitored to prevent bone marrow suppression and gastrointestinal toxicity; peak serum levels (2 hours after an oral dose) should be <100 mg/mL. Persons treated with fluconazole should be monitored for hepatotoxicity, although this toxicity is rare. Management of Treatment Failure Treatment failure is defined as clinical deterioration despite appropriate therapy (assuming increased intracranial pressure is being adequately treated as described previously), the lack of improvement in signs and symptoms after 2 weeks of appropriate therapy, or relapse after an initial clinical response. A repeat lumbar puncture should be performed (if a shunt is not already in place) to ascertain whether or not intracranial pressure has increased. Although fluconazole resistance has been reported with C. neoformans, it is rare. Susceptibility testing is not routinely recommended, and susceptibility techniques have not been standardized for this purpose. The optimal therapy for those with treatment failure is not known. Those who have failed on fluconazole should be treated with amphotericin B with or without flucytosine as indicated previously, and therapy should be continued until a clinical response occurs (BIII). Higher doses of fluconazole in combination with flucytosine also might be useful (BIII). Unlike caspofungin, voriconazole has activity against Cryptococcus spp. in vitro and might be an alternative. Prevention of Recurrence Patients who have completed initial therapy for cryptococcosis should be administered lifelong suppressive treatment (i.e., secondary prophylaxis or chronic maintenance therapy) (AI), unless immune reconstitution occurs as a consequence of ART. Fluconazole (AI) is superior to itraconazole (BI) for preventing relapse of cryptococcal disease and is the preferred drug (361,362). Adult and adolescent patients appear at low risk for recurrence of cryptococcosis when they have successfully completed a course of initial therapy, remain asymptomatic with regard to signs and symptoms of cryptococcosis, and have a sustained increase (i.e. >6 months) in their CD4+ T lymphocyte counts to >100--200 cells/µL after ART. The numbers of such patients who have been evaluated remain limited. On the basis of these observations and inference from more extensive data regarding safety of discontinuing secondary prophylaxis for other opportunistic infections during advanced HIV-1 disease, discontinuing chronic maintenance therapy among such patients is a reasonable consideration (CIII). Certain HIV specialists would perform a lumbar puncture to determine if the CSF is culture-negative and antigen negative before stopping therapy even if patients are asymptomatic; other specialists do not believe this is necessary. Maintenance therapy should be re-initiated if the CD4+ T lymphocyte count decreases to <100--200 cells/µL (AIII). Special Considerations During Pregnancy Diagnosis and treatment for cryptococcosis among HIV-1--infected pregnant women are the same as for nonpregnant women. Considerations about the use of amphotericin B, fluconazole, and itraconazole are the same as those for mucocutaneous and invasive candidiasis (i.e., amphotericin B should be used in the first trimester to avoid the potential for teratogenicity with fluconazole or itraconazole). Flucytosine is teratogenic in rats at high doses, but not at doses similar to human exposure (363). No reports exist about its use in the first trimester of pregnancy in humans. Flucytosine might be metabolized to 5-fluoruracil. It should be used in pregnancy only if clearly indicated. HistoplasmosisEpidemiology Histoplasmosis is caused by the dimorphic fungus Histoplasma capsulatum and occurs in 2%--5% of patients with AIDS who reside in areas in the United States where the disease is endemic (e.g., the Midwest and Puerto Rico) and who are not receiving ART (364--366). In areas where the disease is not endemic, it most often occurs among those who have previously lived in an area where the disease is endemic. Histoplasmosis is acquired by inhalation of microconidia of the mycelial phase of the organism, but reactivation of latent infection might be a mechanism for disease in certain patients. Disseminated histoplasmosis usually occurs among persons with CD4+ T lymphocyte counts <150 cells/µL; localized pulmonary histoplasmosis might occur among persons with CD4+ T lymphocyte counts >300 cells/µL. The incidence of histoplasmosis appears to have declined with the use of potent ART. Clinical Manifestations The most common clinical presentation of histoplasmosis among patients with AIDS is disseminated multiorgan disease. Patients usually have fever, fatigue, and weight loss; respiratory tract symptoms of cough, chest pain, and dyspnea might occur in up to 50% of patients (367). Symptoms and signs might be limited to the respiratory tract for those with higher CD4+ T lymphocyte counts and localized pulmonary histoplasmosis. Septic shock syndrome occurs in <10% of patients. CNS, gastrointestinal, and cutaneous manifestations each occur in <10% of cases, and other sites might be less commonly involved. Diagnosis Detection of Histoplasma antigen in blood or urine is a sensitive method for rapid diagnosis of disseminated histoplasmosis but insensitive for pulmonary infection. Antigen is detected in the urine of 95% and serum of 85% of patients with disseminated histoplasmosis (368) and might be present in bronchoalveolar lavage fluid or CSF of patients with pulmonary or meningeal involvement. Fungal stain of blood smears or tissues also might yield a rapid diagnosis, but the sensitivity is <50%. H. capsulatum can be isolated from blood, bone marrow, respiratory secretions or localized lesions in >85% of cases, but isolation can take 2--4 weeks (367,368). Serologic tests are positive in approximately two thirds of cases but are rarely helpful in the acute diagnosis of histoplasmosis disease. Diagnosis of meningitis poses added difficulties. Fungal stains are usually negative, and CSF cultures are positive in no more than half of cases (369). Antigen or anti-Histoplasma antibodies can be detected in the CSF in up to 70% of cases. Among certain patients, none of these tests are positive, and a presumptive diagnosis of Histoplasma meningitis might be appropriate if the patient has disseminated histoplasmosis and findings of CNS infection not explained by another cause. Treatment Recommendations Patients with severe disseminated histoplasmosis who meet one or more selected criteria (temperature >102oF [>39oC], systolic blood pressure <90 mm Hg, pO2 <70 torr, weight loss >5%, Karnofsky performance score <70, hemoglobin <10 g/dL, neutrophil count <1000 cells/µL, platelet count <100,000 cells/µL, aspartate aminotransferase >2.5 times normal, bilirubin or creatinine >2 times normal, albumin <3.5 g/dL, coagulopathy, presence of other organ system dysfunction, or confirmed meningitis) should be treated with intravenous amphotericin B, either the deoxycholate formulation or liposomal amphotericin B, for the first 3--10 days until they clinically improve (370,371) (AI). In a randomized clinical trial, liposomal amphotericin B was more effective than the standard deoxycholate formulation (371), inducing a more rapid and more complete response, lowering mortality, and reducing toxicity (BI). Intravenous itraconazole 200 mg/day after an initial higher dose induction period might be used for persons who cannot tolerate amphotericin B (BIII). Patients responding well after completion of initial amphotericin B therapy for 3--10 days might be switched to oral therapy with itraconazole capsules to complete 12 weeks of treatment and then placed on maintenance treatment (372) (AII). Itraconazole solution would be logical to use, but no trials document efficacy and tolerability in this setting. Fluconazole 800 mg daily is less effective than itraconazole (373), but is recommended as an alternative if patients cannot tolerate itraconazole (CII). For persons with confirmed meningitis, amphotericin B should be continued for 12--16 weeks, followed by maintenance therapy (AII). Fluconazole has been recommended previously among HIV-1--uninfected persons with meningitis following amphotericin B; however, because of the data documenting efficacy of itraconazole in persons with HIV-1 disease and nonmeningeal histoplasmosis, itraconazole should be used in this setting (AII). Among persons with mild illness, therapy with itraconazole capsules for 12 weeks is recommended (AII). Acute pulmonary histoplasmosis in an HIV-1--infected patient with intact immunity, as indicated by a CD4+ T lymphocyte count >500 cells/µL, might not require therapy and should be managed in a similar way to infection in an otherwise noncompromised host (370) (AIII). Prevention of Recurrence Patients who complete initial therapy for histoplasmosis should be administered lifelong suppressive treatment (i.e., secondary prophylaxis or chronic maintenance therapy) with itraconazole 200 mg twice daily (373) (AI). Certain specialists recommend serum levels be tested to ensure free itraconazole concentrations of at least 1 mg/mL or free plus hydroxylated metabolite of 2 µg/mL. The metabolite also has antifungal activity. Although patients might be at low risk for recurrence of systemic mycosis when their CD4+ T lymphocyte counts increase to >100 cells/µL in response to ART, the number of patients who have been evaluated is insufficient to warrant a recommendation to discontinue secondary prophylaxis in this setting. Special Considerations During Pregnancy Treatment is the same as for nonpregnant adults. Because fluconazole is teratogenic in high doses in animal studies and itraconazole is teratogenic in high doses among rats and mice, as with other invasive fungal infections, amphotericin B should be substituted for itraconazole or fluconazole (if indicated) in the first trimester (BIII). CoccidioidomycosisEpidemiology Coccidioidomycosis is caused by Coccidioides immitis and occurs predominantly in the Southwestern United States where the disease is endemic. However, sporadic cases might be diagnosed in areas where the disease is not endemic as a result of reactivation of previous infection. The incidence of disease in endemic areas was from 2%--5% in the pre-ART era. Increased risk is associated with extensive exposure to disturbed soil. Both localized pneumonia and disseminated infection are usually observed in those with CD4+ T lymphocyte counts <250 cells/µL. The use of ART appears to have reduced the incidence in this patient population. Clinical Manifestations The two most common clinical presentations of coccidioidomycosis are disseminated disease and meningitis. Disseminated disease is associated with generalized lymphadenopathy, skin nodules or ulcers, peritonitis, liver abnormalities, and bone and joint involvement. Localized meningeal disease results in symptoms of lethargy, fever, headache, nausea or vomiting, or confusion and occurs in approximately 10% of patients. Among those with meningeal involvement, CSF analysis typically demonstrates a lymphocytic pleocytosis with CSF glucose levels <50 mg/dL. CSF protein might be normal or mildly elevated. Diagnosis The diagnosis of coccidioidomycosis is confirmed by culture of the organism from clinical specimens or by demonstration of the typical spherule on histopathological examination of involved tissue. Blood cultures are positive in a minority of patients. C. immitis serology is frequently positive among HIV-1--infected patients with coccidioidomycosis and is useful in diagnosis. Complement fixation serology (IgG) is generally positive in the CSF in coccidioidal meningitis. Treatment Recommendations For nonmeningeal pulmonary or disseminated disease, amphotericin B is the preferred initial therapy (374,375) (AII). Data evaluating lipid formulations of amphotericin B are limited such that appropriate dosing recommendations cannot be made. Therapy with amphotericin B should continue until clinical improvement is observed, which usually occurs after administration of 500--1,000 mg. Certain specialists would use an azole antifungal concurrently with amphotericin B (BIII). Fluconazole or itraconazole might be appropriate alternatives for patients with mild disease (374,375) (BIII). Coccidioidal meningitis should be treated with fluconazole, which has been reported to be successful in approximately 80% of patients with C. immitis meningitis (376) (AII). Treatment for patients with meningeal disease requires consultation with a specialist. Intrathecal amphotericin B is the most accepted alternative but is toxic (CIII). Prevention of Recurrence Patients who complete initial therapy for coccidioidomycosis should be administered lifelong suppressive therapy (i.e., secondary prophylaxis or chronic maintenance therapy) using either fluconazole 400 mg daily or itraconazole 200 mg twice daily (AII). Although patients might be at low risk for recurrence of systemic mycosis when their CD4+ T lymphocyte counts increase to >100 cells/µL in response to ART, the numbers of patients who have been evaluated are insufficient to warrant a recommendation to discontinue secondary prophylaxis in this setting. Special Considerations During Pregnancy Coccidioides infections appear to be more likely to disseminate if acquired during pregnancy among HIV-uninfected women, with the risk increasing with increasing gestational age (377). This increased risk might be related to the agonistic effect of estradiol and progesterone, both found at high levels during pregnancy, on the growth of C. immitis (378). The risk for dissemination among HIV-1--infected pregnant women has not been evaluated. Invasive fungal infections should be treated the same in pregnancy as in the nonpregnant woman, with the exception that amphotericin B is the preferred agent in the first trimester because of the potential teratogenic risks of the azoles if efficacy is expected to be superior or similar to that of the azoles (BIII). AspergillosisEpidemiology Aspergillosis, most frequently caused by Aspergillus fumigatus but occasionally by other Aspergillus species, was more common before the advent of potent ART among patients with advanced HIV-1 disease (379). Specific risk factors include neutropenia, low CD4+ T lymphocyte count, use of corticosteroids, exposure to broad spectrum antibacterial therapy, and previous pneumonia or other underlying lung disease. Patients who have had HIV-1--associated aspergillosis diagnosed typically have extremely low CD4+ T lymphocyte counts (i.e., <50 cells/µL), a history of other AIDS-defining opportunistic infections, and are not receiving ART. Clinical Manifestations Two major syndromes have been described among patients with AIDS: respiratory tract disease (either semi-invasive pseudomembranous tracheitis or invasive pneumonitis) and CNS infection occurring as a febrile diffuse meningoencephalitis syndrome with vascular infarction as a central feature (based on the predilection of Aspergillus organisms to invade blood vessel walls). Semi-invasive pseudomembranous tracheitis is associated with fever, cough, dyspnea, stridor or wheezing caused by airway constriction, culminating in airway obstruction if untreated. Endoscopic examination demonstrates a confluent, exudative pseudomembrane adherent to the tracheal wall. Invasive pneumonitis occurs with fever, cough, dyspnea, chest pain, hemoptysis, and hypoxemia; chest radiograph demonstrates either a diffuse interstitial pneumonitis or a localized wedge-shaped dense infiltrate representing pulmonary infarction, related to the predilection of the organisms for invasion of vascular endothelium. Diagnosis A definitive diagnosis requires the presence of relevant clinical signs and symptoms and the histopathologic demonstration of organisms in biopsy specimens obtained from involved sites or from a site that is expected to be sterile (e.g., liver or brain). A presumptive diagnosis of respiratory tract disease can be made in the absence of a tissue biopsy if Aspergillus spp. are cultured from a respiratory sample, a compatible lesion or syndrome is present, and no alternative causative process is identified. Serologic testing is not helpful. Treatment Recommendations The recommended treatment for invasive aspergillosis is voriconazole. Amphotericin B, either conventional or lipid formulations, in doses equivalent to 1 mg/kg body weight/daily of standard amphotericin B is an alternative regimen (AIII). Voriconazole has not been studied in this patient population. Caspofungin is approved for patients failing to tolerate or improve with standard therapy; however, it has not been studied in this patient population. Monitoring and Adverse Events Patients should be monitored for adverse effects related to amphotericin B. Airway obstruction can result from extensive pseudomembrane formation in those with tracheitis. Pulmonary infarction and progressive interstitial pneumonitis can lead to respiratory failure. Management of Treatment Failure The overall prognosis is poor among patients with advanced immunosuppression and in the absence of effective ART. Treatment failure is generally defined as failure to respond to initial therapy or progression of clinical signs and symptoms despite appropriate therapy. No data are available to guide recommendations for the management of treatment failure. If amphotericin B was used initially, substitution with voriconazole might be considered; the alternative approach would be rational for those who began therapy with voriconazole (BIII). Prevention of Recurrence No data are available to base a recommendation for or against chronic maintenance or suppressive therapy among those who have successfully completed an initial course of treatment (CIII). Special Considerations During Pregnancy As with other invasive fungal infections, aspergillosis should be treated the same in pregnancy as in the nonpregnant adult, with the exception that amphotericin B is the preferred agent in the first trimester because of the potential teratogenic risks for the azoles, if efficacy is expected to be superior or similar to that of the azoles (BIII). Cytomegalovirus DiseaseEpidemiology Cytomegalovirus (CMV) is a double-stranded DNA virus in the Herpesvirus family that might reactivate to cause disseminated or localized end-organ disease among patients with advanced immunosuppression who have been previously infected with CMV. The majority of infections derive from reactivation of latent infection. Before potent ART, an estimated 30% of patients with AIDS experienced CMV retinitis some time between the diagnosis of AIDS and death (380--382). Incidence for new cases of CMV end-organ disease have reached <2--3 cases per 100 person-years; for those with established CMV retinitis, recurrences continue at a rate estimated at <25% of the peak during the mid-1990s. Endorgan disease caused by CMV occurs among persons with advanced immunosuppression, typically those with CD4+ T lymphocyte counts <50 cells/µL, who are either not receiving or have failed to respond to ART (380--382). Other risk factors include previous OIs, particularly MAC disease, and high plasma HIV-1 RNA levels (>100,000 copies/mL). Clinical Manifestations Retinitis is the most common clinical manifestation of CMV end-organ disease. CMV retinitis usually occurs as unilateral disease, but in the absence of therapy, viremic dissemination results in bilateral disease in the majority of patients. Peripheral retinitis might be asymptomatic or present with floaters, scotomata, or peripheral visual field defects. Central retinal lesions or lesions impinging on the macula are associated with decreased visual acuity or central field defects. The characteristic ophthalmologic appearance of CMV lesions includes perivascular fluffy yellow-white retinal infiltrates, typically described as a focal necrotizing retinitis, with or without intraretinal hemorrhage, and with little inflammation of the vitreous unless immune recovery with potent ART intervenes (380). Blood vessels near the lesions might appear to be sheathed. Occasionally, the lesions might have a more granular appearance. In the absence of ART or specific anti-CMV therapy, retinitis invariably progresses, usually within 10--21 days after presentation. Progression of retinitis occurs in "fits and starts" and causes a characteristic brushfire pattern, with a granular, white leading edge advancing before an atrophic, gliotic scar. Colitis is the second most common manifestation, occurring in 5%--10% of persons with AIDS and CMV end-organ disease (381). The most frequent clinical manifestations are fever, weight loss, anorexia, abdominal pain, debilitating diarrhea, and malaise. Extensive mucosal hemorrhage and perforation can be life-threatening complications. Esophagitis caused by CMV, which occurs in <5%--10% of persons with AIDS who develop CMV end-organ disease, causes fever, odynophagia, nausea, and occasionally mid-epigastric or retrosternal discomfort (366). CMV pneumonitis is uncommon, but when it occurs, it presents with shortness of breath, dyspnea on exertion, a nonproductive cough, and hypoxemia, associated with interstitial infiltrates on chest radiograph. CMV neurologic disease causes dementia, ventriculoencephalitis, or ascending polyradiculomyelopathy (382). Patients with dementia typically have lethargy, confusion, and fever, but the clinical presentation might mimic that of HIV-1 dementia. CSF generally demonstrates lymphocytic pleocytosis (although a mixture of neutrophils and lymphocytes might be seen), low-to-normal glucose levels, and normal-to-elevated protein levels. Patients with ventriculoencephalitis have a more acute course, with focal neurologic signs, often including cranial nerve palsies or nystagmus, and rapid progression to death. Periventricular enhancement of CT or MRI images is indicative of CMV ventriculoencephalitis rather than HIV-1--related neurologic disease. CMV polyradiculomyelopathy causes a Guillian-Barre--like syndrome characterized by urinary retention and progressive bilateral leg weakness. The clinical symptoms generally progress over several weeks to include loss of bowel and bladder control and to flaccid paraplegia. A spastic myelopathy has been reported and sacral paresthesia might occur. The CSF generally indicates a neutrophilic pleocytosis (usually 100--200 neutrophils/mL and some erythrocytes) accompanied by hypoglycorrhachia and elevated protein levels. Diagnosis CMV viremia can be detected by PCR, antigen assays, or blood culture and is generally detected in end-organ disease, but viremia also might be present in the absence of end-organ disease (383--387). The presence of serum antibodies to CMV is not diagnostically useful. A negative IgG antibody level indicates that CMV is unlikely to be the cause of the disease process being investigated (although rarely, primary CMV infection occurs and is associated with end-organ disease), but certain patients with advanced immunosuppression might serorevert from antibody positive to antibody negative; as a result, a negative CMV IgG antibody test does not definitively eliminate the possibility of CMV disease. The diagnosis of CMV retinitis is generally made on the basis of recognition of characteristic retinal changes observed on funduscopic examination by an experienced ophthalmologist. The demonstration of mucosal ulcerations on endoscopic examination combined with colonoscopic or rectal biopsy with histopathological demonstration of characteristic intranuclear and intracytoplasmic inclusions are required for the diagnosis of CMV colitis (381). The diagnosis of CMV esophagitis is established by the presence of extensive large, shallow ulcers of the distal esophagus and biopsy evidence of intranuclear inclusion bodies in the endothelial cells with an inflammatory reaction at the edge of the ulcer (366,381). Culturing CMV from a biopsy or cells brushed from the colon or the esophagus is not sufficient to establish the diagnosis of CMV colitis or esophagitis because certain persons with low CD4+ T lymphocyte counts might be viremic and have positive cultures for CMV in the absence of clinical disease (366). Diagnosis of CMV pneumonitis should be made in the setting of pulmonary interstitial infiltrates and identification of multiple CMV inclusion bodies in lung tissue, and the absence of other pathogens that are more commonly associated with pneumonitis in this population (385). CMV neurologic disease is diagnosed on the basis of a compatible clinical syndrome and the presence of CMV in cerebrospinal fluid or brain tissue (382,387). Detection of CMV is greatly enhanced by PCR in this setting (383,387). Treatment Recommendations The choice of initial therapy for CMV retinitis should be individualized based on the location and severity of the lesion(s), the level of underlying immune suppression, and other factors such as concomitant medications and ability to adhere to treatment (AIII). Oral valganciclovir, intravenous ganciclovir, intravenous ganciclovir followed by oral valganciclovir, intravenous foscarnet, intravenous cidofovir, and the ganciclovir intraocular implant coupled with valganciclovir are all effective treatments for CMV retinitis (388--393) (AI). The ganciclovir intraocular implant plus oral valganciclovir is superior to once daily intravenous ganciclovir (and presumably to once-daily oral valganciclovir) for preventing relapse of retinitis (388--393) (AI). For this reason, certain HIV specialists recommend the intraocular implant plus valganciclovir as the preferred initial therapy, particularly for patients with immediately sight-threatening lesions (adjacent to the optic nerve or fovea); others prefer oral valganciclovir alone (BII). Among patients with peripheral lesions that are not immediately sight-threatening, oral valganciclovir is preferable to the ganciclovir intraocular implant, intravenous ganciclovir, or intravenous foscarnet (AII) (391) because of its greater ease of administration and lack of surgical or catheter-associated complications. However, any of the treatment regimens can be chosen because epidemiologic studies and clinical trials have not demonstrated substantially reduced rates of loss of visual acuity among patients treated with the ganciclovir implant compared with those treated with systemic therapies (AII) (392,393). Certain clinicians would not treat small peripheral CMV retinitis lesions if ART is to be initiated soon because immune recovery might ultimately control the retinitis. However, immune recovery uveitis might be more common among patients given less aggressive anti-CMV therapy (394--397). Therefore, treatment of CMV retinitis until sufficient immune recovery occurs (i.e., CD4+ T lymphocyte count >100 cells/µL for 3--6 months) is still preferred (AIII). For therapy of colitis or esophagitis, the majority of specialists would treat with intravenous ganciclovir or foscarnet (or with oral valganciclovir if symptoms are not severe enough to interfere with oral absorption) for 21--28 days (BII) or until signs and symptoms have resolved. Certain HIV specialists would withhold therapy unless moderate to severe symptoms justify the use of systemic treatment (BIII) if ART is soon to be initiated or can be optimized. Treatment should be considered for persons with histologic evidence of CMV pneumonitis who do not respond to treatment of other pathogens (AIII). For neurological disease, initiating therapy promptly is critical for an optimal clinical response. Although combination treatment with ganciclovir and foscarnet might be preferred as initial therapy to stabilize disease and maximize response (BII), this approach is associated with substantial rates of adverse effects, and optimal treatment for neurologic disease if ART can be optimized is unknown. Studies are underway to evaluate the utility of pre-emptive therapy with systemic treatment among patients with CMV viremia and no evidence of organ system disease. Until such studies are completed, treatment of CMV viremia in the absence of organ system involvement is not recommended (DIII). No data are available to demonstrate that starting ART among treatment-naïve patients with CMV retinitis would have an adverse effect on retinitis, gastrointestinal disease, or pneumonitis, or worsen immune recovery uveitis if this occurs. Therefore, no reason exists to delay initiation of appropriate ART, which should be administered to those with acute CMV retinitis, gastrointestinal disease, or pneumonitis (BIII). Although, no data indicate that immune recovery inflammatory reactions worsen CMV neurologic disease syndromes, because of the localized morbidity that might occur with such an inflammatory reaction, a brief delay in initiation of ART in this setting until clinical improvement occurs might be prudent (CIII). Monitoring and Adverse Events Management of CMV retinitis requires close monitoring by an experienced ophthalmologist and the primary clinician. Dilated indirect ophthalmoscopy should be performed at the time of diagnosis of CMV retinitis, after completion of induction therapy, 1 month after the initiation of therapy, and monthly thereafter while the patient is on anti-CMV treatment (AIII). Monthly fundus photographs, using a standardized photographic technique that documents the appearance of the retina, provide the optimum method for following patients and detecting early relapse (AIII). Adverse effects of ganciclovir include neutropenia, thrombocytopenia, nausea, diarrhea, and renal dysfunction. Adverse effects of foscarnet include anemia, nephrotoxicity, electrolyte abnormalities, and neurologic dysfunction. Seizures have been reported with both ganciclovir and foscarnet. For patients receiving ganciclovir or foscarnet, monitoring of complete blood counts and serum electrolytes and renal function should be performed twice weekly during induction therapy and once weekly thereafter (AIII). Cidofovir is associated with dose-related nephrotoxicity and hypotony. For patients receiving intravenous cidofovir, blood urea nitrogen, creatinine, and urinalysis should be performed before each infusion; administration of the drug is contraindicated if renal dysfunction or proteinuria is detected. Immune recovery uveitis is an immunologic reaction to CMV characterized by inflammation in the anterior chamber or vitreous in the setting of immune recovery after initiation of ART and is generally observed among those with a substantial rise in CD4+ T lymphocyte counts in the 4--12 weeks after initiation of ART (394--397). Ocular complications of uveitis include macular edema and the development of epiretinal membranes, which can cause loss of vision. Treatment usually requires periocular corticosteroids or short courses of systemic corticosteroids. Estimated response rates are approximately 50%. Management of Treatment Failure For patients without immune recovery after initiation of ART and who are receiving chronic maintenance therapy with systemic anti-CMV drugs, relapse of retinitis is likely to occur over time. Although drug resistance might be responsible for some episodes of relapse, early relapse is most often caused by the limited intraocular penetration of systemically administered drugs (398--400). Because it results in greater drug levels in the eye, the placement of a ganciclovir implant in a patient who has relapsed while receiving systemic treatment (IV ganciclovir or oral valganciclovir) is generally recommended and often will control the retinitis for 6--8 months until the implant requires replacement (401,402) (BIII). Reinduction with the same drug followed by reinstitution of maintenance therapy can control the retinitis, although for progressively shorter periods of time (403), and the majority of specialists recommend this approach for initial treatment of relapsed disease (AII). Changing to an alternative drug at the time of first relapse typically does not result in superior control of the retinitis but should be considered if drug resistance is suspected or if side effects or toxicities interfere with optimal courses of the initial agent (403) (AIII). Combination ganciclovir and foscarnet are generally superior to systemic therapy with either agent alone for patients with relapsed retinitis (403) but is accompanied by greater toxicity; this approach might be considered for patients who are not candidates for other alternatives (BI). Drug resistance occurs among patients receiving long-term therapy (404--406). Reported rates typically are <10% during the first 3 months of therapy but increase to 25%--30% by 9 months of therapy (404--406). Reported rates are similar for ganciclovir, foscarnet, and cidofovir (404,405). Low-level resistance to ganciclovir occurs through mutations in the CMV UL97 (phosphotransferase) gene, and high-level resistance to ganciclovir typically occurs because of mutations in both the CMV UL97 and UL54 (DNA polymerase) genes (407--410). Resistance to foscarnet and resistance to cidofovir each occur because of mutations in the CMV UL54 gene. High-level resistance to ganciclovir is frequently associated with cross-resistance to cidofovir (409) and occasionally to foscarnet (411). Although early relapse is generally not a result of resistance, later relapse often is. Because patients with resistant CMV nearly always have mutations in the CMV UL97 gene, and because a limited number of mutations produce the majority of cases of resistance, resistance testing in peripheral blood using a CMV DNA PCR assay and sequencing for CMV UL97 mutations or using a point mutation assay (412,413) might be reasonable for patients who relapse on therapy. Although this approach also has not been validated, certain specialists would recommend performance of resistance testing using this technique, if available, to guide therapy in those with repeated relapses of CMV disease (CIII). Patients with low-level ganciclovir-resistant isolates in the eye might respond to a ganciclovir implant because of the higher local levels of ganciclovir resulting from this form of therapy. However, patients with high-level ganciclovir resistant isolates typically will not respond and will require a switch to alternative therapy. Repetitive intravitreous injections of fomivirsen can be used for relapsed retinitis (BI) but should be combined with systemic therapy (414) (AI). Prevention of Recurrence After induction therapy, secondary prophylaxis (i.e., chronic maintenance therapy) is recommended for life (90,386--390) (AI), unless immune reconstitution occurs as a result of ART. Regimens demonstrated to be effective for chronic suppression in randomized, controlled clinical trials include parenteral or oral ganciclovir, parenteral foscarnet, combined parenteral ganciclovir and foscarnet, parenteral cidofovir, and (for retinitis only) ganciclovir administration through intraocular implant or repetitive intravitreous injections of fomivirsen (AI). Oral valganciclovir has been approved by FDA for both acute induction therapy and for maintenance therapy, although published data are limited. Repetitive intravitreous injections of ganciclovir, foscarnet, and cidofovir have been effective for secondary prophylaxis of CMV retinitis in uncontrolled case series. Intraocular therapy alone does not provide protection to the contralateral eye or to other organ systems and typically is combined with oral valganciclovir. The choice of a chronic maintenance regimen for patients treated for CMV disease should be made in consultation with a specialist. For patients with retinitis, this decision should be made in consultation with an ophthalmologist and should take into consideration the anatomic location of the retinal lesion, vision in the contralateral eye, the immunologic and virologic status of the patient, and the patient's response to ART. Patients with immediately vision-threatening lesions need prompt anti-CMV therapy because progression of the retinitis can occur during the time in which immune recovery is occurring. Daily oral ganciclovir is less effective than daily intravenous ganciclovir for maintenance therapy (389) and with the availability of oral valganciclovir should no longer be used (DIII) (391). Patients with immediately sight-threatening retinitis still might benefit most from the use of the ganciclovir implant and its superior ability to control retinitis progression (BIII). However, replacement of the ganciclovir implant at 6--8 months might not be necessary for those with sustained immune recovery. If the ganciclovir implant is used, it should be combined with oral valganciclovir until immune recovery occurs (BIII). Chronic maintenance therapy is not routinely recommended for gastrointestinal disease but should be considered if relapses occur (BII). A role for maintenance therapy for CMV pneumonitis has not been established (CIII). Discontinuing secondary prophylaxis (chronic maintenance therapy) should be considered for patients with a sustained (> 6 months) increase in CD4+ T lymphocyte counts >100--150 cells/µL in response to ART (415--420) (BII). Such decisions should be made in consultation with an ophthalmologist and should take into account such factors as magnitude and duration of CD4+ T lymphocyte increase, anatomic location of the retinal lesions, vision in the contralateral eye, and the feasibility of regular ophthalmologic monitoring (BII). All patients who have had anti-CMV maintenance therapy discontinued should continue to undergo regular ophthalmologic monitoring for early detection of CMV relapse and for immune recovery vitritis/uveitis (AIII). Relapse of CMV retinitis occurs among patients whose anti-CMV maintenance therapies have been discontinued and whose CD4+ T lymphocyte counts have decreased to <50 cells/µL (420). Therefore, reinstitution of secondary prophylaxis should occur when the CD4+ T lymphocyte count has decreased to <100--150 cells/µL (AIII). Relapse has been reported among patients whose CD4+ T lymphocyte counts are >100 cells/µL, but such reports are rare. Because of the potential for rapid relapse of retinitis when CD4+ T lymphocyte counts decline and the potential for rapid decline of CD4+ T lymphocyte counts with interruption of ART, patients with immune reconstitution not receiving CMV maintenance therapy should still undergo ophthalmologic monitoring (BIII). Special Considerations During Pregnancy The diagnostic considerations among pregnant women are the same as for the nonpregnant women. Indications for treatment of CMV infection during pregnancy are the same as for those in nonpregnant HIV-1--infected adults (AIII). For retinal disease, use of intraocular implants or intravitreous injections for local therapy should be considered in pregnancy if possible to limit fetal exposure to systemically administered antiviral drugs (CIII). Close ophthalmologic monitoring must be maintained, and systemic therapy should then be added as indicated after delivery. Ganciclovir is embryotoxic among rabbits and mice and teratogenic (i.e., cleft palate, anophthalmia, aplastic kidney and pancreas, and hydrocephalus) in rabbits (421--423). Safe use in human pregnancy after organ transplantation has been reported (421,422). On the basis of very limited data and weighing toxicity of the various drugs, ganciclovir is the treatment of choice during pregnancy (BIII). No experience has been reported with the use of valganciclovir in human pregnancy. Concerns are expected to be the same as with ganciclovir. The fetus should be monitored by fetal movement counting in the third trimester and by periodic ultrasound monitoring after 20 weeks of gestation to look for evidence of hydrops fetalis indicating substantial anemia. Foscarnet is associated with an increase in skeletal anomalies or variants in rats and rabbits. No experience with use early in human pregnancy has been reported. A single case report of use in the third trimester described normal infant outcome (424). Because primary toxicity is renal, monitoring of amniotic fluid volumes by ultrasound is recommended weekly after 20 weeks of gestation to detect oligohydramnios. Cidofovir is embryotoxic and teratogenic (i.e., meningomyelocele and skeletal abnormalities) among rats and rabbits. No experience with use in human pregnancy has been reported. Rarely, ultrasound findings in the fetus (e.g., cerebral calcifications, abdominal and liver calcifications, hydrops, microcephaly, ventriculomegaly, ascites, and echogenic fetal bowel) might indicate the possibility of in utero CMV infection among pregnant women with CMV end organ disease (425). In this case, consideration of invasive testing (i.e., amniocentesis and fetal umbilical blood sampling) must be individualized based on clinical history and serologic findings, gestational age, potential risk for HIV-1 transmission, and maternal preference (426). Referral to a maternal-fetal medicine specialist for evaluation, counseling, and potential further testing is recommended. On the basis of data in HIV-uninfected women, transmission of CMV from mother to infant might occur in utero. However, symptomatic infection in the newborn is usually related to primary CMV infection in the mother during pregnancy, and because >90% of HIV-1--infected pregnant women are CMV antibody positive in the majority of studies, the risk for symptomatic infection in the fetus is low (427--431). Therefore, treatment of maternal CMV infection, if asymptomatic, during pregnancy solely to prevent infant infection is not indicated (DIII). Herpes Simplex Virus DiseaseEpidemiology Infections with human herpes simplex virus type 1 (HSV1) and type 2 (HSV-2) are common, with a seroprevalence among adults of HSV-1 approaching 80% and of HSV-2 among persons aged >12 years in the United States of 21.9% (432). Approximately 95% of HIV-1--infected persons are seropositive for either HSV-1 or HSV-2 (432--434). The availability of potent ART has not had an impact on these data. Clinical Manifestations HSV orolabialis is the most common manifestation of HSV1 infection, presenting with a sensory prodrome in the affected area, rapidly followed by the evolution of lesions from papule to vesicle, ulcer, and crust stages on the lips. Ulcerative lesions are usually the only stage observed on mucosal surfaces. The course of illness in untreated subjects is 7--10 days. Lesions recur 1--12 times per year and are often triggered by sunlight or stress. HSV genitalis is the more common manifestation of HSV2 infection. Perineal lesions on keratinylated skin are similar in appearance and evolution to external orofacial lesions. Local symptoms include a sensory prodrome consisting of pain and pruritis. Ulcerative lesions are usually the only stage observed on vaginal or urethral mucosal surfaces. Mucosal disease is generally accompanied by dysuria, vaginal, or uretheral discharge; inguinal lymphadenopathy, particularly in primary infection, is common with perineal disease (434). In profoundly immunocompromised patients, extensive, deep, nonhealing ulceration of the perineum/buttocks might occur. These lesions have been most often reported in those with CD4+ T lymphocyte counts of <100 cells/µL and also might be more commonly associated with acyclovir-resistant virus. HSV keratitis, neonatal HSV, HSV encephalitis, and herpetic whitlow are similar in presentation and treatment to those diseases observed in HIV-seronegative persons but might be more severe. HSV retinitis occurs as acute retinal necrosis, occasionally in the setting of HSV encephalitis. HSV encephalitis occurs among HIV-1--infected persons, but no evidence indicates that it is more severe or common than among HIV-uninfected persons. Diagnosis HSV infections are usually diagnosed empirically on the basis of characteristic skin, mucus membrane, or ophthalmic lesions. With unusual presentations or lesions that do not respond to therapy, swabs from a fresh lesion can be submitted to the diagnostic virology laboratory for Tzanck smear, viral culture, or HSV antigen detection and subsequent antiviral susceptibility testing if necessary. Treatment Recommendations Orolabial lesions can be treated with oral famciclovir, valacyclovir, or acyclovir for 7 days (AII). Moderate-to-severe mucocutaneous HSV lesions are best treated initially with intravenous acyclovir (435--437) (AII). Patients may be switched to oral therapy after the lesions have begun to regress. Therapy should be continued until the lesions have completely healed. Initial or recurrent genital HSV should be treated with oral famciclovir, valacyclovir, or acyclovir for 7--14 days (AII). Trifluridine is the treatment of choice for herpes keratitis, one drop onto the cornea (437) every 2 hours, not to exceed 9 drops/day; it is not recommended for longer than 21 days (AII). Intravenous acyclovir, 10 mg/kg body weight every 8 hours for 14--21 days, is required for HSV encephalitis (AII). Monitoring and Adverse Events Famciclovir, valacyclovir, and acyclovir might occasionally be associated with nausea, vomiting, and diarrhea. Rarely, patients receiving higher doses of valacyclovir or acyclovir might experience renal dysfunction. For patients receiving high-dose IV acyclovir, monitoring of renal function is recommended at initiation of treatment, and once or twice weekly for the duration of treatment, particularly for those with underlying renal dysfunction or those receiving prolonged therapy. Thrombotic thrombocytopenic purpura/hemolytic uremic syndrome resulting in death has been reported among HIV-1--infected patients treated with high-dose valacyclovir but has rarely been reported at conventional doses among persons with HIV-1 disease (438). Management of Treatment Failure Treatment failure related to resistance to antiviral drugs should be suspected if lesions do not indicate signs of resolution within 7--10 days after initiation of therapy. Among immunocompromised patients with suspected acyclovir-resistant HSV, a lesion culture should be obtained and, if virus is isolated, susceptibility testing performed to confirm drug resistance (437). The treatment of choice for acyclovir-resistant HSV is IV foscarnet (439) (AI). Topical trifluridine or cidofovir also has been used successfully for lesions on external surfaces, although prolonged application for 21--28 days or longer might be required. Prevention of Recurrence Chronic therapy with acyclovir is not required after lesions resolve. However, persons who have frequent or severe recurrences can be administered daily suppressive therapy with oral acyclovir, oral famciclovir, or oral valacyclovir (438,440) (AI). Intravenous foscarnet or cidofovir can be used to treat infection caused by acyclovir-resistant isolates of HSV, which are routinely resistant to ganciclovir (AII). Special Considerations During Pregnancy Diagnosis of mucocutaneous and visceral HSV infections is the same in pregnancy as among nonpregnant adults. Treatment of visceral and symptomatic mucocutaneous HSV infections and suppressive therapy for frequent recurrences should be offered during pregnancy as they would be for nonpregnant women (AIII). Acyclovir is the antiviral drug with the most reported experience in pregnancy and appears to be safe (441). Acyclovir is the first choice for therapy of HSV infections in pregnancy (AIII). Valacyclovir is the prodrug of acyclovir. Although experience with use of this drug in pregnancy is limited, its safety profile is expected to be similar to acyclovir (441--444). Famciclovir was not teratogenic in animal studies but experience with use during human pregnancy is limited. Exposures to this drug during pregnancy should be reported to the Famciclovir Registry (888-669-6682). Because of potential teratogenicity and toxicity, foscarnet should be reserved for severe mucocutaneous or visceral HSV infections that have failed to respond to high dose acyclovir, valacyclovir, or famciclovir. An additional concern with HSV during pregnancy is the potential for transmission to the fetus and neonate. The rate of transmission to the fetus and neonate among HIV-1--infected pregnant women co-infected with HSV is not known. Although isolated cases of in utero transmission with primary infection during pregnancy among HIV-uninfected women have been reported, the predominant risk, regardless of HIV-1 co-infection, is from maternal genital shedding at delivery. Cesarean delivery is recommended for women with a prodrome or visible HSV genital lesions at the onset of labor (425) (BIII). Use of acyclovir in late pregnancy suppresses genital herpes outbreaks and shedding in late pregnancy among HIV-seronegative women and might reduce the need for Cesarean delivery for recurrent HSV (426). However, the safety and efficacy of this strategy has not been evaluated among HIV-1--infected women who are more likely to have antibody to HSV-2 and to have both symptomatic and asymptomatic reactivation of genital HSV (426). Therefore, the use of acyclovir specifically to reduce the need for Cesarean delivery among HIV-1--infected women is not recommended (DIII). Varicella Zoster Virus DiseaseEpidemiology Up to 95% of the adult population is seropositive for varicella zoster virus (VZV), and recurrent disease in the form of herpes zoster occurs in 3%--5% of all adults but becomes more prevalent in the elderly and the immunocompromised host. The incidence of herpes zoster is 15--25 times greater in HIV1--infected persons than in the general population and 3--7 times greater than among the elderly. Zoster among HIV1--infected adults can occur at any CD4+ T lymphocyte count; more advanced immunosuppression might be associated with altered manifestations of VZV infection but does not appear to substantially alter the overall incidence of VZV. Clinical Manifestations Herpes zoster (shingles) might follow a prodrome of pain that resembles a burn or muscle injury in the affected dermatome; skin lesions, which are similar to chickenpox in appearance and evolution, develop in the same dermatome. Extensive cutaneous dissemination and visceral involvement have been reported but are rare. Progressive outer retinal necrosis is a VZV-associated entity that typically occurs among HIV-1--infected persons with CD4+ T lymphocyte counts <50 cells/µL. This rapidly progressive necrotizing herpetic retinopathy is often associated with dermatomal zoster and is characterized by multifocal retinal opacification with little or no ocular inflammation (445,446) and rapid visual loss. Acute retinal necrosis occurs as a peripheral necrotizing retinitis with yellowish thumbprint lesions, retinal vascular sheathing, and vitritis with a high rate of visual loss, often caused by retinal detachment. This syndrome can occur in immunologically normal and immunologically deficient persons. Among patients with HIV-1 infection, acute retinal necrosis can occur at any CD4+ T lymphocyte count, although it more often occurs at higher CD4+ T lymphocyte counts, and progressive outer retinal necrosis more often occurs at lower CD4+ T lymphocyte counts. Chickenpox, the principal clinical manifestation of primary VZV in childhood or adulthood, is uncommon in adults and adolescents with HIV-1 infection. When chickenpox occurs, it begins with a respiratory prodrome, followed by the appearance of pruritic vesiculopapular lesions that are more numerous on the face and trunk than on the extremities. Lesions evolve over a 5-day period through macular, papular, vesicular, pustular, and crust stages. In profoundly immunocompromised hosts, vesicles can persist for weeks and coalesce to form large lesions that resemble a burn. VZV has been associated with transverse myelitis, encephalitis, and vasculitic stroke among HIV-uninfected persons. Anecdotal reports of these syndromes exist among HIV-1--infected patients. Diagnosis Zoster and chickenpox are generally diagnosed empirically on the basis of the appearance of characteristic lesions. When lesions are atypical or the diagnosis is uncertain, swabs from a fresh lesion or a sample of biopsied tissue can be submitted for viral culture or antigen detection. Treatment Recommendations The recommended treatment for localized dermatomal herpes zoster is famciclovir or valacyclovir for 7--10 days (AII). If cutaneous lesions are extensive or if clinical evidence of visceral involvement is observed, intravenous acyclovir should be initiated and continued until cutaneous lesions and visceral disease are clearly resolving (447) (AII). Because of its immunosuppressive effects and the absence of data to support benefit with its use in this patient population, adjunctive corticosteroid therapy to prevent postherpetic neuralgia is not recommended (DIII). Progressive outer retinal necrosis is rapidly progressive and leads to profound loss of vision. Because of the rapidity of disease progression, recommended treatment is high-dose intravenous acyclovir in combination with foscarnet (AIII). Acute retinal necrosis typically responds to IV acyclovir, followed by oral valacyclovir (CIII). Concomitant laser retinal photocoagulation might be needed to prevent retinal detachments. Intravenous acyclovir for 7--10 days is the recommended initial treatment for immunocompromised adults and adolescents with chickenpox (448) (AIII). Switching to oral therapy after the patient has defervesced if no evidence of visceral involvement exists might be permissible (449) (AII). Oral acyclovir is the recommended treatment (20 mg/kg body weight up to a maximum dose of 800 mg four times daily), but valacyclovir or famciclovir would be reasonable alternatives (BII). Monitoring and Adverse Events Recommendations are the same as for HSV. Management of Treatment Failure Treatment failure caused by drug resistance should be suspected if lesions do not indicate signs of resolution within 10 days of initiation of therapy or if they evolve to a verrucous appearance. A lesion culture should be obtained, and if virus is isolated, susceptibility testing performed to confirm antiviral drug resistance and to support the need for intravenous therapy. Among patients with suspected or proven acyclovir-resistant VZV infections, treatment with intravenous foscarnet is the recommended alternative therapy (439) (AI). Prevention of Recurrence No drug has been proven to prevent the recurrence of zoster (shingles) among HIV-1--infected persons. Special Considerations During Pregnancy Diagnosis of zoster and chickenpox during pregnancy is the same as among nonpregnant adults. Treatment of zoster during pregnancy should be the same as for nonpregnant women. Oral valacyclovir therapy is the preferred treatment for HIV-1--infected pregnant women who experience chickenpox during pregnancy (BI). Intravenous acyclovir should be used if parenteral therapy is indicated the same as for nonpregnant adults with varicella (BI). Women should be monitored closely for signs of pneumonitis or other systemic manifestations and hospitalized for observation and potential administration of intravenous acyclovir for any respiratory symptoms or signs of severe disease. HIV-seronegative women with primary VZV infection (i.e., chickenpox) during pregnancy have a 0.4% risk for transmitting infection resulting in congenital varicella syndrome in the infant when infection occurs at or before 12 weeks of gestation. The risk increases to 2.2% with infection at 13--20 weeks, and is negligible after 20 weeks (450). Specific risks among HIV-1--infected women with primary VZV infection during pregnancy have not been reported. Women with primary VZV during the first half of pregnancy should be counseled about the risks and offered detailed ultrasound surveillance for findings indicative of fetal congenital varicella syndrome (450). Provision of varicella zoster immune globulin (VZIG) does not alter the risk of congenital varicella syndrome (450). Infants born to women who have chickenpox anytime from 5 days before through 2 days after delivery should receive VZIG to reduce the severity and mortality rate of neonatal infection acquired during maternal viremia (450) (AII). The maternal care provider should notify the infant's medical provider immediately of the onset of maternal chickenpox during the peripartum period. Human Herpesvirus-8 DiseaseEpidemiology Human herpesvirus-8 (HHV-8) is a transmissible DNA virus with a seroprevalence in the United States of 1%--5%. The seroprevalence is considerably greater among MSM, regardless of HIV-1 infection, and is also much higher in certain Mediterranean countries (10%--20%) and in parts of sub-Saharan Africa (30%--80%). HHV-8 is associated with all forms of Kaposi sarcoma (i.e., classic, endemic, transplant-related, and AIDS-related), and certain rare neoplastic disorders (e.g., primary effusion lymphoma and multicentric Castleman disease). The precise pathogenesis is unclear even though seroconversion to HHV-8 precedes the development of these tumors (451). Patients who are HHV-8 seropositive and have HHV-8 DNA in their peripheral blood have a greatly enhanced risk (approximately ninefold) for experiencing Kaposi sarcoma compared with HHV-8 seropositive men without HHV-8 DNA in their blood (452). The overall incidence of Kaposi sarcoma was as high as 20% among patients with AIDS before the advent of effective ART. However, even before the widespread use of ART, the incidence had declined, which certain specialists attribute to the use of ganciclovir, foscarnet, and cidofovir for the acute and maintenance treatment of CMV disease (based on data demonstrating that these agents inhibit the replication of HHV-8 in vitro) (453--455). Studies indicate that patients receiving ganciclovir or foscarnet (but not acyclovir) have a reduced rate for developing Kaposi sarcoma (390,456--458) or lesion regression after ganciclovir or foscarnet therapy (459,460). Anecdotal reports exist of lesion regression among patients who have been treated with potent ART, and despite the lack of precise epidemiologic studies, the incidence of Kaposi sarcoma has declined dramatically after the introduction of protease inhibitor drugs and ART. Primary effusion lymphoma and multicentric Castleman disease remain rare. Kaposi sarcoma, primary effusion lymphoma, and multicentric Castleman disease are described most frequently among HIV-1--infected persons with more advanced immunosuppression (CD4+ T lymphocyte counts of <200 cells/µL), although they can occur at any CD4+ T lymphocyte count. Clinical Manifestations Because the principal clinical manifestations of HHV-8 infection are neoplastic diseases, the occurrence, diagnosis, and treatment recommendations for these entities are beyond the scope of this report. Diagnosis Routine screening for HHV-8 by PCR or serologic testing is not indicated for HIV-1--infected persons. Treatment Recommendations Although ganciclovir, foscarnet, and cidofovir have in vitro activity against HHV-8, and limited studies indicate these agents might be associated with reduced disease progression or lesion regression, larger and more definitive studies are needed to determine whether antiviral therapy has a useful role in managing HHV-8--associated diseases. Potent ART that suppresses HIV-1 replication reduces the frequency of occurrence of Kaposi sarcoma among HIV-1--infected persons and should be considered for all persons who qualify for such therapy (BII). Prevention of Recurrence Effective suppression of HIV-1 replication with ART among HIV-1--infected patients with Kaposi sarcoma might prevent Kaposi sarcoma progression or occurrence of new lesions and should be considered for all persons with evidence of active Kaposi sarcoma (BII). Special Considerations During Pregnancy The seroprevalence of HHV-8 infection among HIV-1--infected pregnant women varies widely by geographic area, ranging from 1.7% among U.S.-born and 3.6% among Haitian-born women in New York City to 11.6% among pregnant women from four other U.S. cities (461). Pregnancy does not appear to affect the prevalence of antibodies to HHV-8 or the antibody levels (462). HHV-8 seropositivity does not appear to impact pregnancy outcome. Routine screening for HHV-8 by PCR or serology is not indicated for pregnant women. Diagnosis of Kaposi sarcoma or other HHV-8--associated neoplasms in pregnancy should be the same as among nonpregnant women. Recommendations for the treatment of HHV-8 malignancies are beyond the scope of this report. Treatment should be undertaken in consultation with a specialist. Perinatal transmission of HHV-8 occurs but appears to be infrequent. A study of 32 mother-infant pairs indicated that maternal HHV-8 infection might increase the risk for perinatal transmission of HIV-1, although no evidence of HHV-8 infection was identified among HIV-1-infected infants (463). Data indicate increased mortality through 24 months among HIV-1--infected infants born to HHV-8 seropositive compared with HHV-8 seronegative mothers (464--471). Evidence supporting vertical transmission during pregnancy or the intrapartum period includes cases of Kaposi sarcoma developing shortly after birth (464,465), higher risk for transmission with higher maternal antibody titer (and by inference higher HHV-8 viral titers) (466), and detection of HHV-8 DNA by PCR in specimens drawn at birth from infants born to HHV-8 seropositive mothers. The majority of studies demonstrate a rate of persistent antibody positivity in children of 2%--29% by age 4 years with the majority of studies documenting a substantially higher rate of seropositivity among children born to HHV-8 antibody positive compared with antibody negative women (467--471). Progressive Multifocal Leukoencephalopathy Caused by JC VirusEpidemiology Progressive multifocal leukoencephalopathy (PML) is an AIDS-defining neurologic disease caused by the JC polyoma virus. The JC virus is an ubiquitous polyoma virus; the name is derived from the initials of the first patient from whom this virus was isolated. The majority of humans are infected early in life, and 70% of adults have detectable serum antibodies. Clinical Manifestations No known symptoms associated with acute JC virus infection exist. PML is the only known disease caused by the JC virus. This disease has an insidious onset and produces a neurologic syndrome that progresses relatively rapidly over weeks or months, characterized by cognitive dysfunction, dementia, seizures, ataxia, aphasia, cranial nerve deficits, hemiparesis or quadriparesis, and eventually coma. Typical computed tomographic abnormalities include single or multiple hypodense, nonenhancing cerebral white matter lesions, although cerebellum and brain stem are occasionally involved. Diagnosis A confirmed diagnosis of PML requires a compatible clinical syndrome and radiographic findings coupled with brain biopsy demonstrating characteristic pathologic foci of demyelination and oligodendrocytes with enlarged nuclei and basophilic-staining intranuclear material. Whether a brain biopsy will yield information that will alter the clinical course of a patient presenting with a demyelinating disease is a clinical judgement. PCR detection of JC virus DNA in CSF provides supportive diagnostic information in the presence of a compatible clinical syndrome and radiographic findings, and can be used for diagnosis when a brain biopsy is not feasible. Treatment Recommendations No effective therapy for JC virus exists. Randomized clinical trials have evaluated vidarabine and cidofovir; neither is effective in producing clinical improvement and neither is recommended (472,473) (EI). When ART is initiated and CD4+ T lymphocyte counts rise, certain patients will experience neurologic improvement and others might become neurologically stable. However, reports have documented patients experiencing worse neurologic manifestations after initiation of ART. In certain instances, this worsening is caused by an immune reconstitution inflammatory syndrome; other cases represent the natural history of PML. Prevention of Recurrence No role exists for antiviral agents in the prevention of recurrence or progression of PML. Human Papillomavirus DiseaseEpidemiology Human papillomavirus (HPV) infection of the anogenital tract results in a spectrum of disease, ranging from self-limited, transient infection to squamous cell cancer. HPV is the etiologic agent of genital warts and condyloma acuminata. A small number of HPV types (typically HPV types 6, 11, 40, 42, 53, or 54) are associated with warts on the external anogenital skin and types 6 and 11 account for approximately 90% of lesions in the majority of series (474--476). Genital tract HPV infections are thought to be transmitted by sexual contact (476). Lesions sometimes occur at anatomic locations away from sites of direct contact (477). Both the incidence and prevalence of genital warts is increased among patients with HIV-1 infection (478,479). The incidence of genital warts is increased by >10 times among HIV-1--infected women compared with uninfected women (479). In parallel with the increased prevalence of genital tract HPV infection, cervical intraepithelial neoplasia (CIN) and anal intraepithelial neoplasia (AIN), associated with certain HPV subtypes (16, 18, 31, 35 and others) occur with increased frequency among HIV-1--infected women compared with HIV-seronegative women. The relative risk for CIN is 5--10 times higher for HIV-1 seropositive women. Additional factors that contribute to increased risk for CIN include being black, having a history of smoking, younger age at first intercourse, and the number of sex partners. HIV-1--infected women with CIN manifest higher grade lesions than HIV-seronegative women, particularly in the setting of lower CD4+ T lymphocyte counts and higher plasma HIV-1 RNA levels (480--483) and are at higher risk for having HPV-associated lesions elsewhere in the anogenital tract including the vagina, vulva and anus, potentially associated with vaginal intraepithelial neoplasia (VAIN), vulvar intraepithelial neoplasia (VIN), and AIN (484). Women with high-grade CIN or vulvar cancer might have a high incidence of both AIN and invasive anal cancer. MSM have a high prevalence of anal HPV infection and AIN, and those who are HIV-1--infected, particularly those with lower CD4+ T lymphocyte counts, are at even higher risk than those who are not (485--487). In addition to the high incidence of anal HPV infection and AIN among HIV-1--infected men and women, the incidence of anal cancer is higher in these groups than in the general population. Although data are limited, effective ART does not appear to substantially influence the short-term natural history of CIN, AIN, or other forms of intraepithelial neoplasia. Clinical Manifestations The principal manifestation of genital HPV infection is the presence of a cauliflower-like, pedunculated lesion or lesions that might be a few millimeters to as much as 1--2 centimeters in diameter or flat, keratotic plaques and dome-shaped papules (often keratotic). Multiple lesions are usually present and they often occur in clusters. Lesions might occur at multiple sites in the anogenital tract. Certain patients are asymptomatic, although those with perianal lesions might complain about pain on defecation or perianal itching. No characteristic symptoms are associated with CIN. Vaginal bleeding might occasionally accompany cervical or vaginal lesions. Lesions might be visible on the ectocervix, typically at the squamo-columnar junction, during routine pelvic examination. VAIN, VIN, AIN, or frank malignancy might occur with bleeding or itching and lesions might be visible or palpable. Diagnosis HPV disease can be diagnosed by clinical inspection, and further diagnostic testing is not generally required. The entire anogenital tract should be carefully inspected for visual signs of warts. A digital examination of the vulvar, vaginal, and perianal regions and the anal canal should be performed as part of routine evaluation. Digital examination should be performed after collection of a cervical or anal Papanicolou (Pap) smear because lubricant may obscure the interpretation of Pap smears. If uncertainty about the etiology of visible lesions or the presence or absence of high-grade dysplasia or malignancy exists, the diagnosis can be confirmed by a biopsy. Even typical condylomata among HIV-1--infected persons might have foci of high-grade dysplasia. Biopsies of warts should be considered earlier in the evaluation in those with HIV-1 infection than among uninfected persons. Testing for HPV DNA is available, but no clinical indications exist for routine testing of anogenital warts for the presence or type of HPV. Guidelines should be followed for routine Pap smear and colposcopic monitoring to detect dysplasia among HIV-1--infected women (Table 3) (488). If a Pap smear is performed and returns with a cytologic interpretation reporting "atypical squamous cells of uncertain significance" (ASCUS) or "atypical squamous cells-cannot rule out high-grade disease" (ASC-H), an HPV Hybrid Capture™ test can be performed. If the HPV Hybrid Capture™ test reveals an oncogenic HPV type, or if the Pap smear interpretation reports a low-grade squamous intraepithelial lesion (LSIL) or a high-grade SIL (HSIL), colposcopic evaluation and directed biopsy are recommended. Although formal guidelines recommending anal Pap smear screening have not been adopted, certain specialists recommend anal cytologic screening for HIV-1--infected men and women. High-resolution anoscopy (HRA) should be considered if the anal Pap smear indicates ASCUS or ASC-H and should be performed if a person has LSIL or HSIL on anal Pap smear. Visible lesions should be biopsied to determine the level of histologic changes and to rule out invasive cancer. Treatment Recommendations Treatments are available for genital warts, but none is uniformly effective. The rate of recurrence is high with most modalities (489). Data are limited on the response of HIV-1--infected patients to the available treatments for genital warts. In the absence of data specific to the HIV-1--infected population, guidelines for the treatment of sexually transmitted diseases should be followed. Data are insufficient to recommend a single treatment modality for all patients, and more than one treatment option might be required for refractory or recurrent lesions among patients with HIV-1 infection. Patient-applied treatments are generally recommended for uncomplicated external lesions, and consist of the following options (CIII):
Provider-applied treatments are generally recommended for complex or multicentric lesions or those lesions inaccessible to patient-applied treatments (CIII). Options are summarized as follows:
The management of CIN among HIV-1--infected patients should not differ from recently published guidelines (488) (AIII). The majority of specialists recommend observation without specific intervention for CIN 1 unless lesions persist over an 18--24 month period of follow-up, evolve to CIN 2 or worse, or there is poor adherence to routine monitoring. Conventional therapies used for treatment of CIN 2 or 3 include cryotherapy, laser therapy, cone biopsy, and a loop electrosurgical excision procedure (LEEP). LEEP is generally the preferred mode of treatment (BIII). Recurrence rates of 40%--60% after treatment have been reported among HIV-1--infected women undergoing these procedures (497). For AIN, data are insufficient to recommend a specific treatment approach; because the majority of lesions are not visible to the patient, the majority of specialists recommend use of one or more of the provider-applied treatments outlined previously (CIII) (Table 4). Treatment decisions are based on assessment of the size and location of the lesion and the grade of histology. The least aggressive approaches should be tried first whenever possible (CIII). If a lesion is too large or if treatment is expected to produce substantial morbidity, then certain specialists recommend following patients without treatment and periodic examinations to monitor for development of cancer. A study reported a low success rate for surgical fulguration of widespread AIN 2 or 3 among HIV-1--infected persons (498). No indications exist for radiation therapy for patients with AIN in the absence of evidence of invasive cancer (EIII). The results of studies do not indicate that treatment for CIN or AIN should be modified for patients receiving ART. Conversely, no evidence indicates that ART should be instituted or modified for the purpose of treating CIN or AIN (CIII), although limited data indicate that ART might be associated with improved response rates. Monitoring and Adverse Effects As efficacy varies with each of the treatments for genital warts, and recurrences are common, patients should be monitored by physical examination for evidence of recurrence. The major toxicity of podofilox and topical podophyllin is local skin irritation. Also, if podophyllin is applied to a large treatment area, systemic absorption can cause nausea, vomiting, and CNS effects. The major toxicity of imiquimod is inflammation at the application site. The major toxicity of cryotherapy is local pain. The major side effects of surgical treatment for genital warts are local pain, bleeding, and secondary infection. The major adverse events associated with acid cauterization are local pain and irritation or ulceration of adjacent normal skin. Intralesional interferon is associated with systemic toxicities of interferon, including fever, fatigue, myalgia, malaise, depression, and other influenza-like symptoms. For patients with CIN 1 that has not been treated with one of the outlined interventions, Pap smears or colposcopy should be performed every 4--6 months to monitor for persistence or progression of lesions. As the recurrence of CIN and cervical cancer after conventional therapy is increased among HIV-1--infected persons, patients should be carefully followed after treatment with frequent cytologic screening and colposcopic examination when indicated according to published guidelines (498,499). Management of Treatment Failure Treatment failure is defined as the persistence or recurrence of lesions after appropriate therapy. If evidence exists of persistent or recurrent genital warts, re-treatment with any of the modalities previously described should be considered, preferably with an alternative modality to the one that previously failed (AIII). For persistent or recurrent CIN 2 or 3, repeat loop excision or one or more of the other treatment modalities should be considered (AIII) (499). Prevention of Recurrence No indication exists for secondary prophylaxis (chronic maintenance therapy) with any of the conventional modalities to prevent recurrence of genital warts. Patients with CIN should be monitored with frequent cytologic screening and, when indicated, colposcopic examination for recurrent lesions (AI). In a study of HIV-1--infected women treated for high-grade cervical lesions using conventional therapies, low-dose intravaginal 5-fluorouracil (i.e., 2 g twice weekly for 6 months) reduced the short-term risk for recurrence and possibly the grade of recurrence (500). However, clinical experience with this therapy is too limited to provide a recommendation for its use (CIII). Special Considerations During Pregnancy The decision about whether to treat genital warts during pregnancy should be individualized on the basis of the extent of the warts, concurrent symptoms, gestational age, and patient preference (CIII). Podophyllin and podofilox should not be used during pregnancy (EIII). Use of podophyllin has been associated with an increased risk for fetal death in several animal models and case reports in humans, but not with congenital anomalies. No experience with imiquimod in human pregnancy has been reported; therefore, its use in pregnancy is not recommended (DIII). No anomalies have been observed among animals with use during pregnancy. Other topical treatments (e.g., bichloroacetic and trichloroacetic acid) and ablative therapies (i.e., laser, cryotherapy, and excision) can be used during pregnancy. Cervical warts should be biopsied to rule out concomitant dysplasia. Increased bleeding might occur with cervical biopsy during pregnancy. All pregnant women should have a Pap smear at their initial prenatal visit unless a normal cervical cytology result has been obtained within the past year. Cytobrush sampling can be done during pregnancy (501). Pregnant women with abnormal cervical cytology results should undergo colposcopy and cervical biopsy of any abnormalities. Increased bleeding might occur with cervical biopsy during pregnancy. Endocervical curettage should not be done during pregnancy (502,503) (DIII). Repeat cytology with or without colposcopy should be conducted at 34--36 weeks of gestation to rule out progression of dysplasia. Women with any grade of cervical dysplasia can deliver vaginally (if otherwise appropriate based on obstetrical and HIV parameters) with repeat colposcopy and definitive therapy completed postpartum. Women with suspected cervical cancer should be referred to a gynecologic oncologist for definitive diagnosis, treatment, and delivery planning because vaginal delivery is not recommended with invasive cervical cancer (504). Pregnancy appears to increase the rate of detection of genital HPV DNA among HIV-uninfected women and might be associated with an increased frequency and rate of growth of genital warts (505--507). The effect of pregnancy on genital HPV detection among HIV-1--infected women has not been evaluated. Transmission of genital HPV type 6 and 11 from vaginal secretions at delivery is the presumed mechanism of early onset recurrent laryngeal papillomatosis in infants. Although rare, this condition occurs more frequently among infants delivered vaginally compared with those delivered by Cesarean section (505,506--508). No change in obstetrical management is indicated for women with HPV infection unless extensive condylomata are present that might impede vaginal delivery or cause extensive bleeding (509--512). Hepatitis C Virus DiseaseEpidemiology Chronic hepatitis C is caused by hepatitis C virus (HCV), a single-stranded RNA virus. Six distinct genotypes and approximately 50 subtypes have been described. Genotype 1 infection accounts for approximately 75% of all HCV infections in the United States; genotypes 2 and 3 are more prevalent in Western Europe. Both HCV and HIV-1 are efficiently transmitted through large or repeated percutaneous exposure to infectious blood. Numerous studies have documented a high rate of HCV co-infection (50%--90%) among HIV-1--infected injection-drug users and persons with hemophilia (513--516). Other potential modes of transmission of HCV include mother-to-infant (the rate is approximately 5% but increases to 17% if the mother is HIV-1--infected), needlestick, or sexual (515--522). Data from a large cross-sectional analysis of a heterogeneous group of HIV-1--infected persons participating in clinical trials in the United States indicates that 16.1% were HCV co-infected (523). The majority of co-infected persons had a history of injection-drug use. Long-term studies of persons with HCV alone indicate that approximately 2%--20% of those with chronic HCV infection experience cirrhosis within approximately 20 years after acute infection; older age at the time of infection, male sex, and the presence of concomitant alcoholism increase the frequency (524--528). HIV-1 infection appears to speed the rate of progression of chronic hepatitis C to end-stage liver disease (ESLO) to as little as 10 years after exposure (528--534); however, this accelerated progression has not been observed in all studies. Data from a meta-analysis indicate that the average risk for progressive liver disease is 2.9 times higher among HCV/HIV-1 co-infected persons than among persons infected only with HCV (535). Factors that adversely influence disease progression among HCV/HIV-1 co-infected persons include older age, lower CD4+ T lymphocyte count, and a history of alcoholism. Evaluation of liver histology with an established measure (METAVIR scoring system) indicates the presence of more extensive fibrosis as well as a greater rate of fibrosis progression among HCV/HIV-1 co-infected persons than among those with HCV infection alone (532). Whether HCV accelerates progression of HIV-1 disease is unknown (536--538). More recent studies have reported that HCV infection might accelerate progression of HIV-1 infection, although whether HCV co-infection worsens immunologic dysfunction is unknown (530,538--540). Clinical Manifestations Acute hepatitis C is not often recognized because of a high rate of asymptomatic or mildly symptomatic presentation. A limited proportion (<20%) of patients with acute infection have symptoms characteristic of acute hepatitis including low grade fever, mild right-upper-quadrant pain, nausea, vomiting, anorexia, dark urine, and jaundice. Liver transaminases (serum alanine aminotransferase [ALT] and aspartate aminotransferase [AST]) might be elevated. Chronic hepatitis C is often asymptomatic, although complaints of fatigue are common. Serum cryoglobulins are present (60%) but rarely (<5%) cause symptomatic skin, renal, or neurologic manifestations. With progression of liver disease, patients might experience stigmata of portal hypertension including spider angiomata, temporal wasting, splenomegaly, caput medusa, ascites, jaundice, pruritis, and encephalopathy. A small subset of patients experience cutaneous manifestations including leukocytoclastic vasculitis and porphyria cutanea tarda. A rapidly progressive form of hepatitis C, called fibrosing cholestatic hepatitis, has been reported among patients who are immunosuppressed after solid-organ transplantation. This also might occur among HIV-1--infected patients (541). Serum transaminase levels often fluctuate among patients with chronic HCV infection, regardless of HIV-1 co-infection, and long periods of normal serum transaminase levels might occur, although the majority of patients with chronic HCV infection have evidence of liver injury on liver biopsy (542). This injury might occur after years of relatively quiescent infection (543). A weak association between the degree of ALT elevation and the severity of liver injury has been reported, but studies have not been consistent. Diagnosis All HIV-1--infected patients should be tested for evidence of chronic HCV infection. Initial testing for HCV should be performed using the most sensitive immunoassays licensed for detection of antibody to HCV in blood. To confirm the presence of chronic infection, persons positive for antibody to HCV should be tested for HCV RNA by a qualitative HCV RNA assay with a lower limit of detection of <50 IU/mL. Additional, more specific anti-HCV testing by a recombinant immunoblot assay (RIBA) should be performed for patients with a positive anti-HCV result by immunoassay and a negative test for HCV RNA. False negative anti-HCV immunoassay results might occur among HIV-1--infected persons, but this is uncommon with the most sensitive immunoassays (523,544). Seroreversion in patients with low CD4+ T lymphocyte counts has been described (531). If serologic test results are indeterminate, testing for HCV RNA should be performed. Both qualitative and quantitative assays are available for HCV RNA testing. Three diagnostic assays have been approved by FDA for qualitative detection of HCV RNA. Two of the assays use RT-PCR and have a lower limit of detection of 50--100 IU/mL; the third uses transcription mediated amplification and has a lower limit of detection of 10 IU/mL. A single positive qualitative HCV RNA result is sufficient to confirm the diagnosis of active HCV infection, but a negative result cannot exclude viremia because RNA levels might transiently decline below the limit of detection in persons with active infection. A repeat qualitative assay can be performed to confirm the absence of active infection. Quantitative tests for HCV RNA include quantitative RT-PCR or branched DNA (bDNA) signal amplification assays. An HCV RNA standard has been established that permits normalization of viral titers in IUs; these are not indicative of the number of viral particles in a sample. Although the result in IU/mL provides a reasonable estimate of HCV viral load, substantial variability exists among available assays, and if serial values are required to evaluate disease or monitor antiviral therapy, continued use of the same quantitative assay for all assessments is recommended. HCV viral load does not correlate with degree of histologic injury observed on liver biopsy and does not serve as a surrogate for measuring disease severity, but it does provide important prognostic information about the response to antiviral therapy. Quantitative HCV RNA is also useful for monitoring response to therapy. Co-infected persons should be tested for other comorbid liver conditions. Certain specialists recommend screening for hepatocellular carcinoma using alfa-fetoprotein (AFP) and hepatic ultrasound imaging performed at 6-month intervals among HIV-uninfected patients with chronic HCV infection and documented cirrhosis. Although no data are available to evaluate the predictive value of this approach among HIV-1--infected patients, certain specialists recommend that such screening also be considered for co-infected persons with cirrhosis. An abnormal AFP level should prompt further imaging studies to identify focal mass lesions in the liver parenchyma. Numerous imaging modalities are available to evaluate liver parenchymal changes including ultrasonography and scans using single-photon emission computed tomography (SPECT), CT, or MRI technology. Ultrasonography is recommended as the initial test for screening for liver parenchymal changes, cirrhosis, or preliminary detection of mass lesions. Use of SPECT, CT, or MRI scanning should generally be limited to evaluation of hepatic mass lesions among patients with elevated AFP levels and cirrhosis. Hepatic CT without contrast should not be employed because a meta-analysis has documented this to be inferior to biphasic/triphasic CT for identification of small masses in the liver. Testing for ALT levels is the simplest and least expensive test to assess activity of liver disease and should be performed as part of the initial diagnostic evaluation; however, it is nonspecific, and a single abnormal result provides limited useful information. Liver biopsy remains the only definitive test for evaluation of fibrosis stage, and although data among HIV-1--infected patients are limited, fibrosis stage is the most reliable means to assess prognosis and provide information for decisions about the need for initiation of therapy. Therefore, in the absence of a contraindication to liver biopsy, a liver biopsy is recommended for all HIV-1--infected persons with chronic HCV co-infection who are candidates for antiviral therapy, although certain HIV specialists would initiate antiviral therapy without a pre-treatment liver biopsy. Because data about response to antiviral therapy are limited for HIV-1 co-infected patients with HCV genotype 2, 3 or other nongenotype 1 disease, information is limited to base a specific recommendation about whether testing for genotype is useful in this population; however, certain specialists recommend performance of HCV genotype to assist in making a decision to treat chronic hepatitis C. Because up to 80% of patients with HCV genotype 2 or 3 disease respond favorably to antiviral therapy, a decision to treat is more straightforward, and a pretreatment liver biopsy might not be necessary for those with genotype 2 or 3 disease. If performed, liver biopsies should be evaluated by pathologists with training and experience in hepatic histology (545). Complex matrix models using noninvasive test results might reliably separate patients with minimal fibrosis from those with cirrhosis but fail to clearly distinguish intermediate stages of fibrotic disease progression and should not supplant liver biopsy at this time (546). Additional studies evaluating a variety of noninvasive methods for determining stage of fibrosis in the absence of a liver biopsy are ongoing. Complications of percutaneous liver biopsy (i.e., hemorrhage, biliary peritonitis, and pneumothorax) occur at rates of 1--3 per 1,000 cases. Higher complication rates are reported among patients with or without HIV-1 infection who have substantial thrombocytopenia, coagulation defects, or liver lesions with high vascularity. Although these might not be absolute contraindications to liver biopsy, among these patients, transjugular liver biopsy might be the preferred approach (547). Patients with hemophilia should have adequate platelet factor replacement before a liver biopsy is performed (548,549). Treatment Recommendations Because of the scarcity of published experience treating HIV-1/HCV co-infected persons, practice is dictated largely by principles established for the treatment of HIV-uninfected persons. All patients with chronic hepatitis C should be counseled to avoid alcohol consumption because of the potential increased risk for fibrotic progression (532,548,549). Because fulminant hepatic failure from hepatitis A virus infection occurs at increased frequency in persons with chronic liver disease, persons susceptible to HAV should receive 2 doses of HAV vaccine (BIII). HAV vaccine should be administered before the CD4+ T lymphocyte count declines to <200 cells/µL because the response will probably be better (90). In addition, susceptible HIV-1--infected persons at risk for HBV infection should receive the hepatitis B vaccine series. Antiviral treatment should be considered for all patients with chronic hepatitis C infection (AI). Treatment is recommended for patients at increased risk for development of cirrhosis (i.e., those with chronic hepatitis C who have detectable plasma HCV RNA levels on a qualitative assay, liver biopsy histologic findings of portal or bridging fibrosis and at least moderate inflammation and necrosis, and persistently elevated ALT levels >2 times the upper limit of normal) (BI). Although patients with normal or only minimally elevated (<2 times the upper limit of normal) ALT levels are likely to have mild disease, some might progress to advanced fibrosis and cirrhosis. Controversy exists about whether to take a biopsy and treat these patients. Several factors should be considered when making a decision to treat, including genotype, degree of fibrosis, patient motivation, symptoms, severity of other underlying conditions, age, and the need for and the type of concomitant ART. As disease progression is likely to be slow among co-infected patients with mild elevations of ALT and no or minimal fibrosis or inflammatory changes on liver biopsy, these patients might not need treatment and should be monitored periodically with serial determinations of ALT and repeat liver biopsy. The most appropriate intervals to monitor such patients have not been determined. No data are available to evaluate the safety and effectiveness of antiviral treatment of HCV for HIV-1 co-infected patients with advanced fibrosis or compensated cirrhosis, although some specialists would consider treatment for such patients. Treatment with interferon (IFN)-based therapies is relatively contraindicated among patients with decompensated liver disease, indicated by coagulopathy, encephalopathy, ascites, or history of bleeding varices (DIII). Liver transplantation, where feasible, should be the primary treatment option for patients with decompensated liver disease (CIII). However, data about the safety and effectiveness of liver transplantation among HIV-1--infected adults are insufficient to recommend its use outside of clinical studies (550). The goals of antiviral treatment of chronic hepatitis C include eradication of HCV infection, prevention of histologic progression of hepatic fibrosis and, among persons with HCV-related cirrhosis, prevention of hepatic decompensation, hepatocellular carcinoma, and death. Although the goals of therapy might not be achievable in all patients, histologic and clinical benefits of therapy might not be limited just to persons with clearance of virus (551--553). Approved therapies for chronic hepatitis C among HIV-uninfected persons include monotherapy with standard interferons (interferon [IFN] alfa-2a, alfa-2b, or IFN alfacon-1) or pegylated (PEG) IFNs (alfa-2a and alfa 2b) and combination therapy with standard or PEG IFN alfa 2a or alfa-2b plus ribavirin. Among HIV-uninfected patients, the combination of PEG IFN plus ribavirin is associated with substantially higher rates of sustained virologic response compared with standard IFNs alone or with ribavirin. Also among HIV-uninfected patients, ribavirin doses adjusted by weight are associated with improved efficacy and less ribavirin-associated toxicity than fixed ribavirin doses. On the basis of ease of administration (once-weekly injection) and the superior efficacy in HIV-uninfected persons, PEG IFN alfa-2a or -2b plus ribavirin has largely replaced use of standard IFN alfa plus ribavirin for the treatment of chronic hepatitis C infection (554). Retrospective series and limited, uncontrolled, prospective clinical trials demonstrate that IFN alfa-2b plus ribavirin is reasonably well tolerated and might eradicate HCV infection among certain HIV-1-infected patients (555--557). Results from two prospective, randomized, controlled trials comparing PEG IFN alfa-2a plus ribavirin with standard IFN alfa-2a plus ribavirin in HIV-1--infected patients with HCV co-infection demonstrate safety and superior efficacy of PEG IFN alfa-2a plus ribavirin compared with conventional IFN plus ribavirin (558). Approximately one third of those without a virologic response who underwent liver biopsy had histologic improvement in fibrosis, despite the absence of a virologic response in one trial (558). On the basis of these data, PEG IFN alfa-2a 180 mcg administered weekly by subcutaneous injection (or PEG IFN alfa-2b 1.5 mcg/kg) plus oral ribavirin in a dose of 600--1,400 mg daily based on weight is the recommended treatment for chronic hepatitis C among HIV-1--infected persons (AI). Patients with contraindications for the use of ribavirin (e.g., unstable cardiopulmonary disease, pre-existing anemia unresponsive to erythropoietin, or hemoglobinopathy) can be treated with PEG IFN alfa (2a or 2b) monotherapy (AII). However, decreased rates of sustained virologic response are expected among patients not receiving ribavirin. The optimal duration of HCV therapy among HIV-1--infected persons is unknown. While awaiting data from ongoing clinical trials, the majority of specialists follow recommendations for HIV-uninfected persons. The duration of treatment using combination therapy with PEG IFN plus ribavirin is 48 weeks for patients with HCV genotype 1 disease who demonstrate an early virologic response (a decrease of at least 2 log10 in HCV viral load as measured by quantitative HCV RNA levels) during the first 12 weeks of treatment (AI). Patients with genotype 1 disease who fail to achieve an early virologic response by week 12 have limited chance of achieving a sustained virologic response regardless of duration of therapy, and treatment may be discontinued after 12 weeks in such patients (BI). The recommended treatment duration is 24 weeks for HIV-1--infected persons with genotype 2 or 3 disease (BII); certain specialists would treat for 48 weeks for co-infected patients with genotype 2 or 3 disease (554,559,560) (CIII). Preliminary data among HIV-1--infected patients indicate that the HCV virologic response correlates with pretreatment CD4+ T lymphocyte count (i.e., higher response rates have been observed among patients with baseline CD4+ T lymphocyte counts >500 cells/µL) (561). Therefore, treatment for HCV should be considered before a decline in CD4+ T lymphocyte count to <500 cells/µL for patients with HIV-1 co-infection (BIII). Conversely, for HIV-1--infected patients with CD4+ T lymphocyte counts <500 cells/µL, initiation of ART should be considered before treatment for chronic hepatitis C (BIII). Clinical trials evaluating this approach are in progress. Monitoring and Adverse Events Quantitative HCV RNA levels are the best estimate of treatment response. Reliability and value of serial quantitative measurement as a marker of treatment response remains to be determined, particularly in clinical practice settings where variation in specimen handling and shipping might decrease validity of HCV RNA change. A sustained virologic response (SVR) is defined as the absence of detectable HCV RNA, using a qualitative or quantitative HCV RNA assay with a lower limit of detection of 50 IU/mL, at 24 weeks after the end of antiviral treatment. Relapse is defined as the absence of detectable HCV RNA at the end of treatment (ETR) that is not sustained over time. Nonresponse is defined as the absence of an ETR or a SVR. However, even in the absence of a SVR, several studies have demonstrated improved liver histology after completion of a course of antiviral treatment. HIV-1--infected patients should have a quantitative HCV RNA assay performed at the end of 12 and 24 weeks of treatment, and those with undetectable HCV RNA levels should have an HCV RNA assay repeated 24 weeks after completion of therapy. It is reasonable for co-infected patients who achieve a sustained virologic response to undergo serial HCV RNA testing at 6-month intervals for an additional 1--2 years to exclude late virologic relapse (or re-infection with HCV for those at risk for continued exposure). The major toxicities of IFN alfa (PEG or standard) include influenza-like symptoms (e.g., fever, myalgia, headache, and fatigue), neuropsychiatric abnormalities (e.g., depression and cognitive dysfunction), cytopenias (e.g., thrombocytopenia and neutropenia including a reduction in CD4+ T lymphocyte count), retinopathy, neuropathy, and exacerbation of autoimmune disease. Depression might be severe enough to trigger suicide. Depending on the severity of these toxicities and individual patient tolerance, they might be dose-limiting or interfere with the ability to complete a course of treatment. The major toxicities of ribavirin include dose-dependent hemolytic anemia, cough, and dyspepsia. In addition, in vitro data have demonstrated drug-drug antagonism between ribavirin and the anti-HIV pyrimidine nucleoside analogues (e.g., zidovudine, stavudine, zalcitabine, and lamivudine). The clinical significance of these drug-drug interactions has not been determined. In addition, ribavirin might potentiate the intracellular activity of didanosine through inhibition of inosine monophosphate dehydrogenase. Case reports have indicated the interaction of RBV and didanosine might lead to clinically significant inhibition of mitochondrial DNA polymerase gamma, resulting in severe pancreatitis and lactic acidosis in certain patients (561--564). Until further safety data are available, the combination of ribavirin and didanosine is generally contraindicated (DIII). Complete blood counts, a CD4+ T lymphocyte count, and mental health should be evaluated before initiation of anti-HCV therapy, and the therapy should be monitored at regular intervals during treatment. Adverse effects of IFN alfa and ribavirin might be modified by the use of adjunctive agents such as antidepressants (neuropsychiatric), filgrastim (neutropenia) and erythropoietin (anemia). Although available data are insufficient to recommend the routine use of these agents in the management of HCV, their use should be considered on a case-by-case basis. Management of Treatment Failure No recommendations are available for treatment of patients who fail to respond to initial antiviral treatment of chronic hepatitis C. Certain patients might benefit from retreatment with PEG IFN-based regimens depending on their previous response, tolerance, and adherence to and the type of previous therapy (i.e., conventional IFN monotherapy), the potential potency of the new treatment regimen, the severity of liver disease, and viral genotype and other underlying factors that influence response. Limited data in non-HIV-1--infected persons with HCV indicate that 15%--20% of nonresponders treated with conventional IFN formulations alone or in combination with ribavirin will achieve a SVR when re-treated with PEG IFN and ribavirin. Those who achieved a decline in HCV RNA to levels <100,000 IU/mL during initial treatment with IFN monotherapy or those with genotypes 2 or 3 appear to have better response rates to retreatment. Additional studies evaluating retreatment are in progress. Prevention of Recurrence For HIV-1 and HCV co-infected patients, durability of treatment response and the requirement for chronic maintenance therapy to prevent recurrence are unknown. Therefore, no recommendations are available for chronic maintenance therapy in this setting. Special Considerations During Pregnancy Pregnant HIV-1--infected women should be tested for HCV infection if not previously tested to allow appropriate management for them and their infants. Transaminase levels tend to decrease and HCV RNA levels to increase during pregnancy. Transaminases might increase transiently postpartum (565--567). Treatment of chronic hepatitis C during pregnancy is not indicated and is not recommended (DIII). Both IFN and ribavirin are contraindicated in pregnancy. Although IFNs are not teratogenic among rats, mice, or rabbits, they are abortifacient at high doses in monkeys and should not be used in pregnant women because of the direct antigrowth and antiproliferative effects of these agents. Approximately 30 cases of human exposure to IFNs during pregnancy have been reported, about half in the first trimester, without clear adverse effects (568--570). Ribavirin is labeled as FDA category X because of its teratogenicity at low doses in multiple animal species. Defects noted in animals include limb abnormalities, craniofacial defects, exencephaly, and anophthalmia. This drug should not be used during pregnancy (EIII). Women of child bearing potential and men receiving ribavirin should be counseled about the risks and need for consistent contraceptive use during and for 6 months after completion of ribavirin therapy (571). Evaluation, including liver biopsy, can be delayed until >3 months after delivery to allow potential pregnancy-related changes in disease activity to resolve. Hepatitis A and hepatitis B vaccination can be given during pregnancy (572). The risk for perinatal transmission varies from zero among HIV-1-seronegative women with undetectable HCV RNA levels, to 4%--8% among predominantly HIV-seronegative women with detectable HCV RNA, to 22% among HIV-1--infected women (573--576). The risk for perinatal transmission of HCV is consistently higher among HIV-1--infected compared with HIV-seronegative women, potentially related to higher HCV RNA levels in HIV-1--infected women or concurrent injection drug use. Perinatal transmission of HCV in both HIV-seronegative and HIV-1--infected women also is potentially related to higher HCV RNA levels, although this finding has been inconsistent (573). Mother-to-child transmission of HIV-1 also might be more frequent among HCV co-infected women compared with HIV-1--infected women without concomitant HCV infection (577). Mode of delivery and breast feeding do not appear to influence HCV transmission in HIV-seronegative women, but elective Cesarean delivery might be protective against transmission of HCV among HIV-1--infected women (573,574,576). The adjusted odds ratio for perinatal transmission of HCV with scheduled Cesarean delivery among HIV-1 infected, HCV seropositive women was 0.36 (0.2--0.8) compared with other modes of delivery in one large study; however, this study did not control for concomitant perinatal transmission of HIV-1 (576). Hepatitis B Virus DiseaseEpidemiology Hepatitis B virus (HBV) is the leading cause of chronic liver disease worldwide (578,579). In developed countries, HBV is transmitted primarily through sexual contact and injection-drug use. Even though risk factors are similar, HBV is transmitted more efficiently than HIV-1 (578--580). Although up to 90% of HIV-1--infected persons have at least one serum marker of previous exposure to HBV (581,582), only approximately 10% have chronic hepatitis B, as evidenced by the detection of hepatitis B surface antigen (HBsAg) in the serum persisting for a minimum of 6 months (583,584). HIV-1 infection is associated with an increased risk for the development of chronic hepatitis B after HBV exposure (584--586). Limited data indicate that co-infected patients with chronic hepatitis B infection have higher HBV DNA levels and are more likely to have detectable hepatitis B e antigen (HBeAg) (587,588), accelerated loss of protective hepatitis B surface antibody (anti-HBs), and an increased risk for liver-related mortality and morbidity (589). Clinical Manifestations Although certain patients are asymptomatic, symptoms of acute HBV infection include fatigue, right-upper-quadrant abdominal pain, nausea, vomiting, fever, and arthralgias followed by jaundice. Although persons with chronic hepatitis B infection might have nonspecific symptoms such as fatigue and right-upper-quadrant abdominal pain, chronic hepatitis B is often clinically inapparent until the onset of ESLD manifested as ascites, coagulopathy, caput medusa, palmar erythema, jaundice, hepatomegaly, splenomegaly, variceal bleeding, or hepatic encephalopathy. Ancillary manifestations of chronic hepatitis B disease also include polyarteritis nodosa, glomerulonephritis, and vasculitis. Diagnosis All HIV-1--infected persons should be tested for HBV (90). The optimal testing strategy for co-infected persons has not been determined. Testing for HBsAg, hepatitis B core antibody (anti-HBc), and hepatitis B surface antibody (anti-HBs) is recommended because this strategy will detect the majority of persons with chronic hepatitis B and those who need vaccination. Serum HBV DNA has been detected in certain persons without HBsAg in whom anti-HBc was the only serum marker of infection (590,591). The interpretation of an isolated anti-HBc is difficult both because false-positive tests for anti-HBc occur and because the clinical significance of anti-HBc alone or with low levels of HBV DNA, even in those with elevated ALT levels, is not known (590--593). Chronic hepatitis B is defined as detection of HBsAg for >6 months. Patients with chronic HBV infection should be tested for HBeAg and antibody to HBeAg (anti-HBe). Severity of liver disease should be assessed initially and at least every 6 months with ALT, albumin, prothrombin time, platelet count, complete blood count, and bilirubin. Transient or persistent elevations in liver transaminases might occur just before loss of HBeAg, on discontinuation of anti-HBV therapy, in association with lamivudine resistance, with hepatotoxicity from anti-HIV therapy or other drugs, or with the acquisition of another hepatitis virus infection such as HAV, HCV, or hepatitis delta virus (HDV) (594--597). Certain specialists also would obtain a test to quantify the circulating HBV DNA in patients diagnosed with chronic hepatitis B on the basis of serologic testing (598). Several assays for HBV DNA are available, but results are not interchangeable. HBV DNA levels are usually high in chronic infection (108--1010 copies/mL of blood); however, available data indicate that HBV DNA levels do not predict progression of liver disease or response to therapy in a manner analogous to plasma HIV-1 RNA levels (598). Patients with chronic hepatitis B are at increased risk for hepatocellular carcinoma (HCC). In HIV-seronegative patients certain specialists recommend monitoring patients with chronic hepatitis B every 6--12 months with an AFP level or ultrasound of the liver, especially if the patient is in a high-risk group (i.e., age >45 years, cirrhosis, or a family history of HCC) (598,599); however, the effectiveness of this screening strategy has not been determined. Among HIV-1--infected patients, the risk for and natural history of HBV-related HCC have not been studied; therefore, the optimal HCC screening method and interval are not known. Until more data become available, it seems reasonable to consider periodic (every 6--12 months) AFP and ultrasound screening among patients with persistent HBsAg, especially those in a group at high risk (598). Liver biopsy remains the only definitive test to assess the grade (necroinflammatory activity) and stage (degree of fibrosis) of liver disease. The rate of progression of chronic hepatitis B disease among patients with HIV-1 co-infection has not been studied, and the optimal indications for liver biopsy are not known; however, because fibrosis grade and stage are the most reliable means to assess prognosis and provide information for decisions about the need for initiation of therapy (598), in the absence of a contraindication, the majority of specialists recommend a liver biopsy for all HIV-1--infected persons with chronic HBV co-infection who are candidates for antiviral therapy. Certain HIV specialists would initiate therapy for chronic hepatitis B without a pretreatment liver biopsy. Treatment Recommendations All patients with chronic hepatitis B disease should be advised to avoid or limit alcohol consumption because of the effects of alcohol on the liver (AIII). In addition, they should be counseled about the risk for household, sexual, and needle-sharing transmission and the need for such contacts to receive hepatitis B vaccine. Because fulminant hepatic failure from HAV infection occurs at increased frequency among persons with chronic liver disease, persons susceptible to HAV should receive 2 doses of hepatitis A vaccine (BIII). HAV vaccine should be administered before the CD4+ T lymphocyte count declines to <200 cells/µL because the response is likely to be better (90,600). The goals of anti-HBV therapy are to reduce HBV-related morbidity and mortality. Surrogate endpoints include sustained suppression of HBV DNA, prevention of liver disease progression, and clearance of HBeAg; treated patients rarely become HBsAg-negative as HBV reservoirs generally are not sufficiently reduced by available anti-HBV therapy. Limited data indicates that any treatment reduces the risk for HCC. Antiviral treatment is recommended for patients who have actively replicating virus in blood (as defined by a positive HBeAg or HBV DNA levels >105 copies/mL) and liver disease as indicated by either an elevated serum ALT (at least 2 times the upper limit of normal) or histopathologic evidence of moderate liver disease activity and/or fibrosis on liver biopsy. The response to therapy is poor for those with a pre-treatment ALT level <2 times the upper limit of normal and therapy should generally be deferred for such patients (DIII). However, ALT levels fluctuate widely in persons with chronic hepatitis B, and the long-term pattern is more useful than an isolated value in patient management. Certain specialists recommend treatment of those with advanced fibrosis or cirrhosis on liver biopsy with any detectable HBV DNA level provided other causes for chronic liver disease have been eliminated. No preferred treatment can be uniformly recommended for all HIV-1 co-infected persons with chronic hepatitis B. Therapy should be individualized, taking into account patient-specific considerations. Because of limited data about the safety and efficacy of chronic hepatitis B treatment among HIV-1--infected persons, patients should be encouraged to enroll in clinical trials. IFN-alfa 2a and 2b, administered in subcutaneous doses of 5 MU daily or 10 MU 3 times per week, are approved for the treatment of chronic hepatitis B disease among HIV-uninfected persons but not among HIV-1--infected patients. Approximately one third of HIV-seronegative patients will clear HBeAg with either of these IFN regimens (598,601), and the response is durable among 80%--90% of persons followed for 4--8 years (602). Among HIV-infected persons with chronic hepatitis B, PEG IFN alfa 2a appears to be superior to standard interferon (603). If either standard or pegylated interferon is used for treatment among HBeAg-positive patients, 16--24 weeks of therapy is recommended (BII); for HBeAg negative patients, who respond less well, a minimum of 12 months and possibly longer is recommended (604) (BIII). Patients who have a substantial decrease (certain specialists suggest >2 log10 copies/mL) or clearance of HBV DNA in response to IFN-alfa 2a or 2b at week 16 but have persistent HBeAg also might be candidates for longer term treatment of 12 months or longer (605); however, data are insufficient to make a firm recommendation in HIV-1--infected patients (601,606--608). Certain specialists recommend that IFN alfa be used in HIV-1 co-infected patients who are candidates for treatment of chronic hepatitis B disease but not HIV-1 (CIII). This strategy preserves lamivudine or tenofovir for later treatment of HIV-1 and avoids certain potential complications of ART. IFN-alfa should not be used among patients with decompensated liver disease (EII). Studies of PEG IFN-alfa among HIV-uninfected patients with chronic hepatitis B are in progress, and it will probably become the preferred IFN formulation. For HIV-1--infected persons who are ART-naïve and require ART, lamivudine 150 mg twice daily is commonly used for treatment of chronic hepatitis B, because of its relative safety, anti-HIV activity, wealth of data about its use among HIV-1--infected persons, and the potential toxicity associated with IFN-alfa (BIII). Lamivudine should be used together with other antiretroviral drugs in a fully suppressive ART regimen. Because of the high rate of development of HBV resistance to lamivudine monotherapy, certain specialists further recommend the use of lamivudine in combination with either adefovir or tenofovir, although data are limited to support this approach (CIII). Seroconversion of HBeAg (loss of HBeAg, accompanied by development of HBe antibody) occurs in 22% of HBeAg-positive HIV-1--infected patients with chronic hepatitis B who are treated with lamivudine for 1 year (609). In HIV-seronegative patients, HBeAg seroconversions are sustained among approximately 80% of patients if lamivudine is continued several months after seroconversion. On the basis of limited data on the duration of treatment, HBeAg-positive, HIV-1/HBV co-infected patients who become HBeAg-negative and anti-e--positive on lamivudine therapy should be treated for a minimum of 1 year or at least 6 months beyond HBeAg seroconversion (BIII). Among HIV-seronegative, HBeAg-negative patients with chronic hepatitis B who are treated with lamivudine, ALT and HBV DNA levels might decline, but high rates of relapse have been reported when therapy is stopped (610). Therefore, the optimal duration of treatment of HBeAg-negative patients, whether HIV-1 infected or not, is unknown (CIII). The combination of lamivudine and IFN does not appear to be superior to either medication alone (611,612), and is not recommended (DII). Adefovir dipivoxil, 10 mg daily, has no anti-HIV activity and is unlikely to select for HIV-1 resistance; therefore, it is an appropriate alternative to IFN-alfa for co-infected patients who require treatment for chronic hepatitis B but do not yet require ART (CIII). However, the long-term safety of adefovir has not been established in HIV-1-infected persons. Tenofovir, 300 mg daily, has similar in vitro anti-HBV activity to adefovir, and expanding human data indicate it is also active against lamivudine-resistant and wild-type HBV. Although tenofovir is not approved for use in the treatment of HBV infection and data are sparse in HIV-1/HBV co-infected patients, certain specialists consider tenofovir to be the optimal choice for persons who need treatment for both HIV-1 infection and chronic hepatitis B (in conjunction with a fully suppressive ART regimen) (CIII). Until long-term data are available that demonstrate the absence of HBV resistance to tenofovir, it might be prudent to use tenofovir in combination with lamivudine (CIII). Tenofovir, if used for treatment of HBV in patients receiving ART, should be added as a single agent for this purpose only if plasma HIV-1 RNA levels are undetectable to avoid selection pressure that engenders drug resistance (CIII). If therapy is indicated for HIV-1 infection but not for chronic hepatitis B, certain specialists would withhold tenofovir, if possible, to allow for its future use for treatment of chronic hepatitis B (CIII). Emtricitabine (200 mg once daily) is also active against HBV replication and could potentially be substituted for lamivudine; however, data are limited for its use for this indication. It is not active against lamivudine resistant HBV. Famciclovir is less active than lamivudine against HBV and is not active in lamivudine-resistant HBV; therefore, its use is not recommended (613--615) (DII). For HBV treatment-naïve patients who require treatment of both HIV-1 infection and chronic hepatitis B, many specialists would recommend use of an ART regimen that includes either lamivudine or emtricitabine along with either adefovir or tenofovir. However, combination therapy for treatment of HBV in this population is not yet supported by data (CIII). Among patients infected with HBV, HCV, and HIV-1, consideration of the need for ART should be the first priority. If ART is not required, the treatment of HCV should be considered before HBV treatment because IFN therapies for HCV also might treat HBV (CIII). If IFN-based therapy for HCV has failed, treatment of chronic hepatitis B with nucleoside or nucleotide analogs can be considered (CIII). Monitoring and Adverse Events A virologic response is defined as a substantial (certain specialists recommend >2 log10 copies/mL) decrease in HBV DNA and loss of HBeAg at the end of treatment. A sustained virologic response is defined as suppression of HBV DNA (level not defined) and loss of HBeAg sustained for >6--12 months after the end of treatment. Among HIV-uninfected persons, the response rates to IFN-alfa or lamivudine-containing regimens are >50% in patients with ALT levels >5 times the upper limit of normal and 20%--35% among patients with ALT levels between 2--5 times the upper limit of normal. Patients for whom therapy is not initiated should be monitored regularly for changes in ALT levels (e.g., every 4--6 months). Other markers of treatment success include improvement in liver histology, normalization of hepatic transaminases, and in those with loss of HBeAg, the development of HBe antibody. Sustained loss of HBsAg is considered by some to be a complete response (604). Although a decline in HBV viral load correlates with response, no threshold HBV viral load has been established that clearly defines a virological response. Side effects of IFN-alfa include influenza-like symptoms and fatigue, which can be reduced by premedication with acetaminophen or a nonsteroidal medication. Other common side effects include weight loss, alopecia, thrombocytopenia, anemia, leukopenia (decreased total CD4+ T lymphocyte count but not percentage), depression, and autoimmune disorders. Hypo- or hyperthyroidism, which is often irreversible, might occur 3--6 months after initiation of therapy with IFN-alfa. As a result, serum TSH level should be monitored at baseline and periodically (e.g., every 3 months) for the duration of treatment. Adefovir causes renal tubular disease and renal excretion of carnitine in a substantial proportion of patients at higher doses, but these side effects are uncommon at the 10 mg/day dose. Substantial renal toxicity with tenofovir has not been reported, although isolated cases of increased serum creatinine or renal tubular dysfunction have been observed. Because of the potential for overlapping toxicities and their similar structure, tenofovir and adefovir should not be used in combination. When anti-HBV therapy with lamivudine, adefovir, or tenofovir is initiated, whether for the purpose of treating chronic hepatitis B or for the treatment of HIV-1 infection, discontinuation is associated with a flare of liver disease in approximately 15% of cases, with loss of the benefit accrued from previous anti-HBV treatment (616). Certain specialists recommend that when anti-HBV therapies are initiated, they should be continued unless contraindicated or unless the patient has been treated for >6 months beyond loss of HBeAg positivity. However, the risks and benefits of this practice are unknown. If anti-HBV therapy is discontinued and a flare occurs, reinstitution of anti-HBV therapy is appropriate because it can be potentially life saving (BIII). Management of Treatment Failure The rate of development of lamivudine resistance is approximately 20% per year among HIV-1/HBV co-infected persons treated with lamivudine (617). Among HIV-1--infected patients who have been on lamivudine and are candidates for treatment of chronic hepatitis B, certain specialists recommend use of adefovir or tenofovir (CIII). How long lamivudine should be continued beyond initiation of a new treatment is unknown (617--621). For HIV-1--infected persons previously treated with a lamivudine-containing ART regimen, uncontrolled data indicate that the combination of adefovir with continued lamivudine has substantial antiviral effect even in the presence of lamivudine-resistant HBV (622). Certain specialists use adefovir to treat chronic hepatitis B among HIV-1--infected patients who have had an inadequate response to a course of lamivudine therapy as evidenced by high plasma HBV DNA levels or persistent serum HBeAg (CIII). Whether lamivudine should be continued (or restarted) if not needed as part of the antiretroviral regimen is unknown. Although data are sparse and the drug is not approved for this indication, certain specialists would recommend tenofovir to treat chronic hepatitis B among HIV-1--infected patients who require ART and remain HBeAg-positive or have high levels of circulating HBV DNA despite >12 months of lamivudine (CIII). Whether lamivudine should be used (or restarted) in such patients is unknown. Flares of liver disease have been reported with development of resistance to lamivudine. If this occurs, addition of tenofovir or adefovir might be life-saving (CIII). HBV DNA testing might be useful in this setting because increasing levels are associated with emergence of lamivudine resistance or relapse, and stable levels should suggest an alternative cause of acute deterioration. ESLD among HBV and HIV-1 co-infected patients is managed as it is in HIV-seronegative patients. IFN is contraindicated in ESLD, but limited data indicate that lamivudine and adefovir (and probably tenofovir) can be safely used (617--619). Liver transplantation has been performed with limited success among selected patients with HBV and HIV-1 infection. If a patient is thought to be a candidate for liver transplantation, early consultation with a transplant center should be obtained because transplantation does not cure HBV infection and adequate post-transplant treatment is required (BIII). Prevention of Relapse and Recurrence Among HIV-seronegative, HBeAg-negative patients with chronic hepatitis B who are treated with lamivudine, ALT, and HBV DNA levels might decline, but high rates of relapse have been reported when therapy is stopped (610). Therefore, the optimal duration of treatment of HBeAg-negative patients, whether HIV-1 infected or not, is unknown (CIII). No known effective means exist to prevent recurrence or flares of chronic hepatitis B. Special Considerations During Pregnancy All pregnant women should be screened for HBsAg. Treatment of symptomatic acute HBV infection during pregnancy should be supportive with special attention given to maintaining blood glucose levels and normal clotting status. Risk for preterm labor and delivery might be increased with acute HBV infection. Treatment of chronic HBV infection is generally not indicated in pregnancy (DIII). If antiretrovirals are administered to the mother to prevent HIV transmission, caution should be used in selecting agents like lamivudine or tenofovir that also suppress HBV and may cause hepatitis flare when discontinued. Hepatitis A vaccination, indicated for persons with chronic hepatitis B, can be given during pregnancy. Infants born to HBsAg-positive women should receive hepatitis B immune globulin and hepatitis B vaccine within 12 hours of birth. The second and third doses of vaccine should be administered at 1 and 6 months of age, respectively (AI). This regimen is >95% effective in preventing HBV infection in these infants. Postvaccination testing for anti-HBs and HBsAg should be performed at age 9--15 months because of the infant's on-going exposure to HBV. If treatment for chronic hepatitis B disease is necessary, lamivudine is the preferred agent because it is not teratogenic in animals or based on human experience including >1,000 first trimester exposures reported to the Antiretroviral Pregnancy Registry (621--623). Lamivudine should only be used in HIV-1--infected pregnant women as part of a fully suppressive ART regimen. Limited information is available about adefovir. It is embryotoxic in mice and caused neonatal thymic lymphoid tissue destruction with use in later pregnancy in mice. No reports of its use in human pregnancy are available. Cases of exposure during pregnancy should be reported to the Antiretroviral Pregnancy Registry (800-258-4263; email: registry@pharmaresearch.com or http://www.apregistry.com). Limited information is available about tenofovir. No birth defects have been seen in studies of rats, rabbits, and monkeys. Decreased fetal weights and increased bone porosity were seen in monkeys after high dose exposure in utero. Nineteen cases of first trimester exposure in humans without birth defects have been reported (621). Cases of exposure during pregnancy should be reported to the Antiretroviral Pregnancy Registry (800-258-4263; email: registry@pharmaresearch.com or http://www.apregistry.com). Geographic OIs of Special ConsiderationPenicilliosisEpidemiology Penicilliosis is caused by the dimorphic fungus, Penicillium marneffei, which is endemic in Southeast Asia (especially Northern Thailand and Southern China) (624--626). Disseminated penicilliosis is the clinical manifestation for 14% of patients with AIDS in northern Thailand (625). International travel requires increased awareness and recognition of penicilliosis and its treatment. The majority of cases of penicilliosis are observed in patients with low CD4+ T lymphocyte counts, usually <50 cells/µL (625). The infection is associated with a high mortality rate if timely treatment with appropriate antifungal drugs is not administered. Clinical Manifestations Penicilliosis is a systemic disease that commonly occurs with fever, skin lesions, weight loss, and bone marrow, lymph node, and hepatic involvement. The skin lesions consist of a generalized papular rash; some of the papules might have central umbilication resembling molluscum contagiosum. Cutaneous penicilliosis lesions commonly appear on the face, ears, extremities, and occasionally the genitalia. Patients with hepatic penicilliosis have fever, abdominal pain, hepatomegaly, and a marked increase in serum alkaline phosphatase levels. Diagnosis The diagnosis is based on isolation of the fungus from blood or other clinical specimens or by histopathologic demonstration of organisms in biopsy material. Fungal cultures demonstrate characteristic features that include a flat green surface and underlying deep red coloring. An early presumptive diagnosis can be made several days before the results of fungal cultures are available by microscopic examination of Wright stained samples of skin scrapings, bone marrow aspirate, or lymph-node biopsy specimens. Many intracellular and extracellular basophilic, spherical, oval and elliptical yeast-like organisms can be seen, some with clear central septation, which is a characteristic feature of P.marneffei. Treatment Recommendations The recommended treatment is amphotericin B in a dose of 0.6 mg/kg body weight/day administered intravenously for 2 weeks, followed by oral itraconazole solution in a dose of 400 mg/day for a subsequent duration of 10 weeks (627) (AII). ART should be administered in accordance with standards of care in the community; consideration should be given to simultaneous administration of treatment for penicilliosis and initiation of ART to improve outcome (CIII). Management of Treatment Failure Alternative treatment options for penicilliosis are not available. For those who fail initial therapy, the approach to treatment should consist of re-initiating parenteral amphotericin B followed by another course of oral itraconazole, coupled with optimizing ART, addressing obstacles to adherence, avoiding adverse drug interactions, and ensuring that adequate absorption and serum concentrations of itraconazole are achieved (AIII). Prevention of Recurrence Relapse is common in the absence of chronic suppressive therapy. All patients who successfully complete treatment for penicilliosis should be administered secondary prophylaxis (chronic maintenance therapy) with oral itraconazole in a dose of 200 mg/day (628) (AI). Special Considerations During Pregnancy Invasive fungal disease should be treated the same in pregnancy as in the nonpregnant adult with the exception that amphotericin B is the preferred agent in the first trimester because of the potential teratogenic effects of azoles. LeishmaniasisEpidemiology Leishmaniasis is caused by Leishmania spp., obligate intracellular protozoa. The organisms survive and replicate in intracellular vacuoles within macrophages. The Leishmania genus has traditionally been differentiated into multiple species, which cause cutaneous, mucosal, or visceral diseases (629,630). Leishmaniasis is endemic in 88 countries in the world. An estimated 12 million cases have been reported worldwide with an incidence of 1.5--2.0 million new cases annually. Co-infection with Leishmania and HIV-1 has been reported in at least 28 countries. Leishmaniasis among persons with HIV/AIDS has been reported primarily from Spain, Italy, France, Portugal, and other countries bordering the Mediterranean, and Central and South America, and South India, although the overall incidence has decreased substantially in developed countries with the introduction of ART (631,632). Disease occurs primarily among those with advanced immunosuppression with low CD4+ T lymphocyte counts, usually <100 cells/µL (629,630). Evidence indicates that after primary infection, Leishmania remain viable among healthy persons for long periods, leading to a susceptible population when immunosuppression intervenes. Primary leishmaniasis is spread almost exclusively by sand flies of the genus Phlebotomus or Lutzomyia; however, in the Mediterranean basin and in Southern Europe, HIV-1 and Leishmania co-infections have been reported predominantly in males and in association with injection-drug use, suggesting that Leishmania might also be acquired by needle sharing. Clinical Manifestations Depending on the persons infected and the species of Leishmania, leishmaniasis can occur in four different syndromes: localized cutaneous, diffuse cutaneous, mucosal, and visceral disease. The most common clinical presentation of leishmaniasis in persons with AIDS is a disseminated visceral disease syndrome (70%). In Mediterranean countries, visceral leishmaniasis among HIV-1--infected patients is in general similar to that observed among non-HIV--infected populations (633). The most common clinical and laboratory findings are fever (80%), splenomegaly (65%), hepatomegaly (63%), and pancytopenia (73%). Splenomegaly is less frequent among HIV-1--infected patients (633). Among those with more profound immunosuppression, atypical manifestations, with involvement of the upper and lower gastrointestinal tract, lung, pleural and peritoneal cavities and skin is common (633--635). In geographic locations other than the Mediterranean basin, clinical manifestations might include unusual nonulcerative cutaneous lesions that mimic Kaposi sarcoma or the more common nodular diffuse form of leishmaniasis. Disfiguring mucosal lesions that are associated with anergy to Leishmania antigens and a negative leishmanin skin test reaction have been observed among persons with AIDS, unlike mucosal lesions in immunocompetent persons that are associated with strong DTH responses (635). Diagnosis Demonstration of characteristic amastigote forms of Leishmania in tissue biopsy specimens (e.g., scrapings, aspirates, other specimens by histopathology, cultures, and smears) from cutaneous or mucosal lesions is the standard for diagnosis of cutaneous leishmaniasis among HIV-1 co-infected patients (630). The diagnosis of visceral leishmaniasis among patients with hepatosplenomegaly is also made by the demonstration of amastigote forms in buffy-coat smear preparations, cultures from the peripheral blood, and smears or cultures from bone marrow or spleen aspirates. Other methods useful for demonstrating Leishmania in the blood of co-infected patients include detection of Leishmania nucleic acid by PCR amplification and xenodiagnosis using colonized sand flies (636--638). Antileishmanial antibodies against Leishmania antigens are of high diagnostic value among immunocompetent patients and can be detected by various serological methods (630). However, among HIV-1 co-infected patients, serologic tests are often negative. The use of recombinant antigen (e.g., rK39) in ELISA assays might increase sensitivity. Also, immunoblotting with Leishmania infantum soluble antigen has been successful in detecting specific antileishmanial antibodies in up to 70% of European patients. Negative DTH responses to leishmanin skin tests are frequently observed in persons with HIV-1/Leishmania co infection, particularly in those with profound immunosuppression and cutaneous anergy to other antigens. Treatment Recommendations Pentavalent antimony, 20 mg/kg body weight/day, administered by intravenous or intramuscular routes, is the initial treatment of choice for leishmaniasis both for cutaneous or visceral disease in many parts of the world (639,640) (AII). The duration of treatment ranges from 3--4 weeks depending on the initial response (633,639--641) (CIII). Antimonials suppress Leishmania infection by decreasing the parasite burden in infected macrophages but do not eradicate infection, and relapses commonly follow cessation of therapy among immunosuppressed patients with AIDS. Patients with visceral leishmaniasis might have severe neutropenia and might benefit from a short course of recombinant human (rHu) granulocyte macrophage colony stimulating factor (GM-CSF) 5 mg/kg body weight/day administered subcutaneously during the initial 5 days of treatment (642) (CII). Amphotericin B is an effective but less extensively evaluated alternative treatment for leishmaniasis (639, 643--645) (AII). Both the conventional and lipid complex or liposomal encapsulated formulations of amphotericin B appear to have similar efficacy compared with pentavalent antimonials (639,640,643--645) (AII). The lipid complex or liposomal preparations are generally better tolerated, and might be preferable to conventional amphotericin B in this setting (644--646) (BII). The optimal amphotericin B dosage has not been determined. Reported regimens include amphotericin B 0.5--1.0 mg/kg body weight/day IV (maximum: 50 mg/day) to achieve a total dose of 1.5--2.0 g (639,641,643) (BII) or lipid complex or liposomal preparations 2--5 mg/kg body weight/day over 10 consecutive days (640,644--646) (BII). If lipid complex or liposomal preparations are used, a higher daily dosage is recommended (639,644,645) (BII). Pentamidine isethionate, 3--4 mg/kg body weight/day administered as a single IV dose infused over at least 60 minutes, at intervals of three times per week for 3--4 four weeks, is a second-line alternative for treatment of leishmaniasis (641,643) (BII). The following regimens have also been reported to have activity in the treatment of visceral leishmaniasis: allopurinol 20 mg/M2 in three doses, alone or combined with pentavalent antimony or imidazoles (CIII), imidazoles such as ketoconazole (400--600 mg/day) or itraconazole (400 mg/day) (CIII); and IFN-gamma as adjunctive therapy for severe or refractory cases of visceral leishmaniasis (647) (CIII). Evidence indicates that HIV-1 co-infection alters T helper cytokine responses to Leishmania (630,633). Poor clinical response to antileishmanial chemotherapy has been reported among co-infected patients who have high plasma HIV-1 RNA levels. Data further indicate that patients receiving ART who present with visceral leishmaniasis have better outcomes than those not receiving ART (631). As such, strong consideration should be given to initiation or optimization of ART among patients who present with leishmaniasis (CIII). Monitoring and Adverse Events Patients receiving pentavalent antimonials should be monitored closely for adverse reactions, which are frequent and vary from mild phlebitis to death. Overall, at a dose of 20 mg/kg bodyweight/day, >60% of patients might have one or more of the following reactions: local pain at the site of injection, thrombophlebitis, anorexia, myalgia, arthralgia, abdominal pain, elevation of liver transaminases, amylase or lipase, and in some patients, clinical pancreatitis. Occasional electrocardiographic changes might be observed (e.g., prolonged QT intervals and T wave inversion). Rarely, arrhythmias and sudden death have occurred (639,640). Patients treated with amphotericin B should be monitored for dose-dependent nephrotoxicity, electrolyte disturbances, and infusion-related adverse reactions, which might be ameliorated by pre-treatment with acetaminophen, diphenhydramine, or limited doses of corticosteroids (CIII). Previous fluid expansion with colloidal fluids might help reduce the risk for nephrotoxicity during treatment (CIII). Management of Treatment Failure For patients who fail to respond to initial therapy or experience a relapse after initial treatment, a repeat course of the initial regimen or use of one or more of the recommended alternatives for initial therapy as outlined above should be considered (CIII). The response rate for retreatment appears to be similar to that for initial therapy although certain patients might evolve to a chronic disease state with serial relapses despite aggressive acute and maintenance therapies (CIII). Although data to support its use among HIV-1--infected persons are limited, miltefosine might be an alternative oral agent for use as salvage therapy in countries outside the United States (CIII). The drug is approved and available in India and registration in Europe is pending. The adult dose is 100 mg daily for 4 weeks. Cure rates in HIV-seronegative patients are reported at approximately 95%. A phase II trial from India indicated that miltefosine was as effective as amphotericin B for the treatment of visceral leishmaniasis in HIV-seronegative patients (648). Gastrointestinal side effects are the most common adverse effects but rarely limit treatment. Prevention of Recurrence Among patients with visceral leishmaniasis who are not receiving or responding to ART, the risk for relapse at 6 and 12 months, in the absence of secondary prophylaxis (chronic maintenance therapy), is 60% and 90%, respectively (630, 649). Therefore, secondary prophylaxis with pentavalent antimony, amphotericin B, or pentamidine, administered at least every 2--4 weeks, is recommended, particularly for those with CD4+ T lymphocyte counts <200 cell/µL (630,633, 646,649--651) (AII). Relapse after discontinuation of secondary prophylaxis or maintenance therapy for leishmaniasis is uncommon among patients who respond to ART and maintain a CD4+ T-lymphocyte count >200 cells/µL (652), although relapse might be more common among those with visceral leishmaniasis, even with CD4+ T lymphocyte counts >200 cells/µL and undetectable plasma HIV-1 RNA (653). Although data are insufficient to provide a recommendation, discontinuation of secondary prophylaxis after successful treatment of leishmaniasis might be considered after a sustained (i.e., >3--6 months) increase in the CD4+ lymphocyte count to levels >350 cells/µL after initiation of ART (652,653) (CIII). Daily allopurinol, in a dose of 300 mg three times daily, used for maintenance therapy is less effective than monthly pentavalent antimony and is not recommended (649) (DIII). Special Considerations During Pregnancy Diagnostic considerations are the same among pregnant women as in nonpregnant adults. Labeling for pentavalent antimony compounds (sodium stibogluconate available in the United States through CDC and meglumine antimoniate) states that they are contraindicated among pregnant women, although various compounds were not teratogenic among chickens, rats, or sheep (654--656). A single case report of use of meglumine antimoniate in the second trimester of human pregnancy reported a good outcome for mother and infant (657,658). Because of concerns about toxicity and lack of experience with use of pentavalent antimony compounds in human pregnancy, amphotericin B would be the first choice for therapy of visceral leishmaniasis in pregnancy if the Leishmania species causing infection is expected to be responsive to amphotericin B (658) (AIII). Pentamidine would be the second choice, and antimony compounds reserved for infections not responsive to the other two agents (AIII). Perinatal transmission of Leishmania spp. occurs rarely; eight documented cases have been reported (659). No data on the risk for transmission of Leishmania spp. among HIV-1--infected pregnant women are available. ParacoccidioidomycosisEpidemiology Paracoccidioidomycosis is caused by Paracoccidioides brasiliensis, a dimorphic fungus that exists in a mycelial phase in the soil and as budding yeast in infected tissue. Paracoccidioidomycosis is the most prevalent endemic mycosis in Central and South America. An estimated 10 million persons are infected. Infection of the susceptible host is presumed to occur after inhalation of the fungus in the mycelial phase (660--663). Relatively few cases of paracoccidioidomycosis in association with HIV-1 infection have been reported. One reason for this is that TMP-SMX prophylaxis for P. jiroveci pneumonia also appears to be effective in preventing clinical disease caused by P. brasiliensis (660--663). However, other factors, such as lack of intersection of the HIV-1 epidemic with areas where the disease is endemic for P. brasiliensis, confusion of the diagnosis with P. jiroveci pneumonia, and use of azole antifungals for oropharyngeal candidiasis, also might have reduced the apparent number of cases. Clinical Manifestations On the basis of a retrospective review of 27 cases, the manifestations of paracoccidioidomycosis among patients with HIV-1 infection are protean (661). Skin lesions, adenopathy, mucosal lesions, and pulmonary infiltrates, all associated with fever and other constitutional symptoms, are frequent. Diagnosis The diagnosis is based on histological identification of the organism or its growth from involved tissue. Yeast forms in tissue typically form a wheel-house pattern because of the radial budding of daughter yeast forms from a mother cell. Paracoccidioidal serology might be useful, although it appears to be positive less frequently among HIV-1--infected persons than among immunocompetent patients. Newer assays (e.g., genomic identification using PCR and antigen detection) are promising. Treatment Recommendations No published randomized clinical trials for the treatment of paracoccidioidomycosis exist. The majority of specialists recommend amphotericin B for initial therapy in severe cases (BII), but the efficacy of other agents (e.g., TMP-SMX and azole antifungals) might be comparable. In particular, single-arm studies of itraconazole, 100--200 mg daily, ketoconazole 200--400 mg, and sulfonamides have demonstrated activity in immunocompetent hosts (BII). Fluconazole is associated with a higher failure rate even at doses up to 600 mg daily and is not recommended (662,663). Potent ART should be administered in accordance with standards of care in the community (AIII). Management of Treatment Failure In the absence of any clinical trials to indicate approaches to the treatment of patients who fail to respond or who relapse after initial treatment, consideration should be given to retreatment with amphotericin B or to the use of azole antifungals (CIII). Prevention of Recurrence Secondary prophylaxis (i.e., chronic maintenance therapy) to prevent relapse should be considered for patients with AIDS and CD4+ T lymphocyte counts of <200 cells/µL, although no data indicate appropriate regimens in this setting. ART should be optimized. Special Considerations During Pregnancy Invasive fungal infections should be treated the same in pregnancy as among the nonpregnant woman. Amphotericin B is the preferred agent in the first trimester because of the potential teratogenic risks for the azoles, if efficacy of amphotericin is expected to be similar to that of azoles (BIII). IsosporiasisEpidemiology Isosporiasis is caused by Isospora belli, a protozoan belonging to the family Apicomplexa, commonly referred to as coccidia. Infection results from the ingestion of sporulated oocysts in contaminated food or water. Infection occurs worldwide, but the prevalence of infection is higher in tropical and subtropical regions. Infection can occur in both immunocompetent and immunocompromised hosts. Isospora belli infections have been observed among different immunocompromised patients and among patients with AIDS (664--666). Clinical Manifestations The most common clinical manifestation of disease is diarrhea, which in patients with AIDS can be similar to that observed with cholera and associated with severe dehydration. Blood is not present in the feces. Systemic symptoms of fever, headache, malaise, abdominal pain, vomiting, and weight loss are also common. Infection primarily involves the small intestine. Colitis is not generally observed. Diagnosis The diagnosis of isosporiasis can be made by stool examination for ova and parasites. Oocysts are ovoid in shape and are 23--36 by 12--17 mM in size. Isospora oocysts autofluoresce a blue-green color under an epifluorescence microscope, enhancing their detection in wet mount preparations. The organisms also stain red with the same modified acid-fast technique used for diagnosis of cryptosporidiosis. No commercial antigen-detection systems have been developed. Schizonts, merozoites, macrogamonts, microgamonts, microgametes, and oocysts can be demonstrated in enterocytes in biopsies of the small or large intestine. Extraintestinal infections with tissue cyst-like stages have been demonstrated in lymph nodes adjacent to the intestine in patients with AIDS. Treatment Recommendations Fluid support should be offered if the diarrhea has resulted in dehydration (AIII). Malnutrition and wasting should be treated with nutritional supplementation (AIII). The drug of choice for therapy is trimethoprim (160 mg) and sulfamethoxazole (800 mg) administered four times a day for 10 days (665) (AII). Doses of oral trimethoprim (320 mg) plus sulfamethoxazole (1,600 mg) taken twice a day for 10--14 days is as effective and might be associated with improved adherence and tolerability (AIII). Treatment with TMP-SMX results in clearance of parasites, decreased volume of diarrhea, and decreased abdominal pain within a mean of 2.5 days after initiation of treatment. No effective alternative treatment is available for patients unable to tolerate sulfonamides. Several agents have been used with anecdotal success. Pyrimethamine used alone in doses of 50--75 mg/day appears comparable to treatment with trimethoprim and sulfamethoxazole (666) (BII). When pyrimethamine is used, it should be administered with folinic acid (5--10 mg/day) to prevent bone marrow suppression (BII). Ciprofloxacin and other fluoroquinolones have demonstrated activity against other Apicomplexa in animal studies and might be second-line alternatives for treatment of isosporiasis (BII). In a limited, randomized clinical trial comparing ciprofloxacin with TMP-SMX among HIV-1--infected patients with isosporiasis, all patients treated with TMP-SMX cleared the organism and had cessation of diarrhea within a median of 2 days; ciprofloxacin was effective in 83% of patients with a median time to cessation of diarrhea of 4.5 days (667). Treatment with other anti-protozoal agents (e.g., metronidazole, tinidazole, quinacrine, and furazolidone) is probably of limited value and is not recommended (DIII). Immune restoration after of ART among patients with AIDS is associated with more rapid resolution of symptoms and fewer relapses. Therefore, ART is recommended as part of the treatment for patients with isosporiasis (AIII). Management of Treatment Failure Treatment failure is defined as persistence or worsening of diarrhea and systemic symptoms after 5--7 days of appropriate treatment. Retreatment with a second-line alternative agent might result in improvement in those who fail initial therapy (BIII). Macrolide antibiotics have marginal efficacy in treating I. belli enteritis (CII). Spiramycin (1.5 g twice daily) and roxithromycin (2.5 mg/kg body weight every 12 hours) have been effective in a limited number of patients with AIDS and chronic refractory isosporiasis (668,669). Diclazuril (200--300 mg/day for 7 days), nitazoxanide (500 mg twice a day for 7--10 days), and albendazole coupled with ornidazole were effective in limited numbers of patients with AIDS and I. belli diarrhea and might be tried among patients unresponsive to (or intolerant of) TMP-SMX (670--672) (CII). Prevention of Recurrence Infections tend to be chronic and relapsing, particularly in patients with AIDS and advanced immunosuppression. Treatment is usually effective in controlling symptoms, but recurrences are common after treatment is stopped, probably because the agents used to treat the infection are not active against the extra-intestinal tissue cyst stage of the parasite. Patients with CD4+ T lymphocyte counts <200/mL should receive secondary prophylaxis (chronic maintenance therapy) with trimethoprim (320 mg) and sulfamethoxazole (1,600 mg) once daily or three times a week (AII). Pyrimethamine, 25 mg per day, also has been used successfully for secondary prophylaxis following primary isosporiasis (BII). Although not evaluated in any clinical trial or observational cohort setting, it is likely, as with other similar opportunistic infections, that secondary prophylaxis can be safely discontinued after an increase in CD4+ T lymphocyte counts to levels >200 cells/µL sustained for at least 3--6 months following initiation of ART (BIII). Special Considerations During Pregnancy The incidence, clinical manifestations, and course of I. belli infection do not appear to differ with pregnancy. Diagnosis and therapy should be the same as among nonpregnant women (673,674). Chagas DiseaseEpidemiology American trypanosomiasis, or Chagas disease, is an anthropozoonosis caused by Trypanosoma cruzi, a flagellated protozoan transmitted to humans and mammals by a group of haematophagous reduviid insects. T. cruzi causes a lifelong chronic bloodstream infection in vertebrate hosts, including humans (675--678). Chagas disease vectors have been reported in the Americas from 42ºN to 46ºS, and the disease is distributed from the southern United States to the southern regions of Argentina and Chile. When reduviid insects bite the vertebrate host's skin to take a blood meal, T. cruzi parasites are deposited with the insect's feces and penetrate through the skin defect into the host. Humans also might acquire trypanosomiasis by blood transfusion, transplacentally, from an infected transplanted organ, or from laboratory accidents (675--678). In 1990, Chagas disease affected 16--18 million persons. Approximately 45,000 deaths annually were attributed to Chagas' disease in the Americas, and approximately 7.2% of the population of Argentina was chronically infected, 22% of Bolivia, 4.3% of Brazil, and 10% of Chile. Since then, the Southern Cone Initiative to interrupt transmission of Chagas disease has reduced the incidence of the disease by 70% in the Southern Cone countries. An estimated 50,000--100,000 persons in the United States are infected with T. cruzi. In humans, T. cruzi infection is followed by an acute illness with moderate to high levels of parasitemia which, after a period of a few months, is followed by a lifelong chronic infection, characterized by low-grade and intermittent parasitemia in which tissue parasites are scarce and difficult to demonstrate. All patients with chronic infection are potentially able to transmit Chagas disease through triatomid insect bites, pregnancy, blood transfusion, or organ donation. Among patients with HIV-1 infection, reactivation of chronic, latent T. cruzi infection can be triggered by profound immunosuppression (679--682). The epidemiology of T. cruzi infection among persons co-infected with HIV-1 in areas where the disease is endemic has not been well-characterized. Clinical Manifestations Chagas disease can be divided into two stages: acute and chronic. The acute phase of Chagas disease, usually observed among children, begins shortly after infection and lasts 1--2 months. This stage of the disease is often asymptomatic, although fever, malaise, anorexia, induration, and lymphadenitis around the inoculation site (chagoma) or periocular edema (Romaña sign) might be observed. Generalized lymphadenopathy and splenomegaly also might occur. Severe illness with cardiac failure or meningoencephalitis occurs in a limited proportion of patients. Acute infection is characterized by relatively high level parasitemia. The acute illness of Chagas disease usually resolves spontaneously, and although the infection does not resolve spontaneously, the patient enters an asymptomatic indeterminate phase of the illness. After one or two decades, 10%--30% of infected patients experience chronic cardiac and/or digestive tract disease. The clinical features of immunosuppression-induced reactivation of Chagas disease among patients with HIV-1 infection differ from that of chronic infection among immunocompetent patients, with the most overt difference being the high frequency of CNS involvement, with attendant high morbidity and mortality. Neurological signs and symptoms are the predominant clinical findings among HIV-1--infected patients with reactivation of T. cruzi infection (679--682). The most common presentation of this form of disease is a severe multifocal or diffuse acute meningoencephalitis with necrosis and hemorrhage associated with a substantial number of amastigotes in tissue. Although most patients have a single supratentorial lesion, some demonstrate multiple lesions in both supratentorial and infratentorial regions. Many of these patients also have detectable peripheral parasitemia. The clinical manifestations of perinatally acquired T. cruzi in HIV-1 co-infected infants have not been well described. Of those reported, the majority have had serious meningoencephalitis syndromes (683,684). Diagnosis Chagas disease should be considered in the differential diagnosis of CNS mass lesions and cardiac disease (arrhythmias or heart failure) among patients with HIV-1 infection in areas where the disease is endemic. The imaging pattern of brain chagoma is similar to that of cerebral toxoplasmosis although chagomas tend to be larger than Toxoplasma lesions. MRI and CT imaging indicate hypodense lesions, which enhance with gadolinium. A definitive diagnosis is established by brain biopsy or identification of the parasite (or its products) in tissue or blood. Preliminary results of tests to identify T. cruzi using a PCR amplification assay of DNA are promising. Direct tests for identifying T. cruzi microscopically are useful during the acute stage and in reactivation of chronic infection (e.g., in the setting of HIV-1 infection) because in these phases, relatively large numbers of parasites circulate in the bloodstream. Blood concentration techniques, such as capillary centrifugation (microhematocrit test) can improve sensitivity (685,686). In blood, T. cruzi sediments are seen just above the buffy coat. Centrifugation of CSF also can be employed among patients with suspected CNS Chagas disease. Parasites also might be observed in lymph nodes, bone marrow, pericardial fluid, and CNS mass lesions. Indirect tests such as xenodiagnosis (recovering the organism after inoculation of laboratory-raised insect vectors) (686) or hemoculture (culture in liquid medium) are somewhat more sensitive than the direct methods, but might take 2--8 weeks to become positive. They are useful in the chronic stages of T. cruzi infection, when the level of parasitemia is low. Serological tests to detect the IgG antibody responses to T. cruzi infection are useful for diagnosis of disease in chronically infected patients, to screen blood donors, and for seroepidemiological studies. The techniques used are indirect hemagglutination, direct agglutination, complement fixation, indirect immunofluorescence, and ELISA (687). In the United States, multiple serologic tests are licensed for diagnosis, but no test is licensed for screening blood donors. Detection of IgM antibodies is not sensitive, even during the acute stage of infection. A serologic diagnosis of Chagas disease should not be relied on unless at least two different types of serological tests for T. cruzi antibodies are positive. Although all of these tests are reasonably sensitive and specific, both false-positive and false-negative reactions have been reported. For that reason, the diagnosis of Chagas disease should not be discarded based on negative serologic tests if the patient comes from a region where the disease is endemic and has clinical findings compatible with Chagas disease. In this instance, direct parasitologic testing (e.g., microscopic examination of brain tissue and/or demonstration of parasitemia) is the best diagnostic strategy. Neonates born to mothers with chronic T. cruzi infection will have positive antibody tests yet might not be infected; parasitologic tests and repeat antibody testing at 6 and 12 months are recommended in this instance (683). Treatment Recommendations Treatment for Chagas disease is uniformly effective among patients with chronic stage disease; however, the available agents are toxic, and consultation with a specialist should be sought. Benznidazole, 5--8 mg/kg body weight/day for 30--60 days, is the initial treatment most commonly recommended (AIII). Nifurtimox, 10mg/kg body weight/day, is an alternative (BIII). Limited data are available evaluating the efficacy of these agents among HIV-1--infected patients with Chagas disease. Neither drug is licensed in the United States; however, nifurtimox is available from CDC under an investigational protocol. Although no data are available specifically to address this question, treatment of acute Chagas' disease is likely to be more effective than treatment of patients with late-stage complications. The potential impact of immune reconstitution caused by ART on HIV-1-related Chagas disease remains to be established; however, it seems likely that maintaining normal immune function will decrease the frequency of reactivation of T. cruzi, as it has with other OIs. As such, initiation or optimization of ART should be considered for patients undergoing treatment for Chagas disease, but information is limited about drug interactions between agents used to treat Chagas disease and available antiretroviral agents (CIII). <Monitoring and Adverse Effects Patients undergoing treatment should be closely monitored because both benznidazole and nifurtimox are toxic. Benznidazole causes peripheral neuropathy, rash, and granulocytopenia. Nifurtimox causes anorexia, nausea, vomiting, abdominal pain and weight loss, restlessness, seizures, and peripheral neuropathy. The adverse effects of both drugs wane when the drugs are discontinued. Management of Treatment Failure Although no data are available among HIV-1--infected persons, certain specialists recommend retreatment with benznidazole or nifurtimox for patients who fail to respond or relapse following initial therapy (CIII). Prevention of Recurrence Patients with HIV-1-infection are potentially at risk for recurrent or relapsing clinical manifestations because of intermittent reactivation of chronic infection. The drugs are only partially effective in the chronic stage of disease, are suppressive rather than curative, and probably require lifelong administration to prevent relapse in the setting of continued immunosuppression. Precise doses and regimens have not been described (CIII). Whether secondary prophylaxis or chronic maintenance therapy should be used routinely among HIV-1--infected patients with latent Chagas disease is unclear. Whether chronic maintenance therapy can be safely discontinued for persons on ART who have a sustained increase in CD4+ T lymphocyte count to levels >200 cells/µL is uncertain. Special Considerations During Pregnancy The seroprevalence of T. cruzi infection among pregnant women in areas where the disease is endemic in Latin America ranges from as high as 50% in urban areas to 81% in rural areas (687). In the United States, seroprevalence data are limited, but one study of 3,765 pregnant women in Houston, Texas, confirmed antibody to T. cruzi in 0.4% of Hispanic women and 0.1% of non-Hispanic women (688). No data are available on the prevalence of T. cruzi antibodies among HIV-1--infected pregnant women in the United States. Congenital infection with T. cruzi might increase the risk for spontaneous abortion, stillbirth, and low birthweight (689,690). Congenital Chagas disease in newborn infants ranges from subclinical to life-threatening with severe neurological and cardiac disease. No data are available to evaluate whether the combination of HIV-1 infection and T. cruzi infection increases the risk for adverse pregnancy outcomes. Diagnosis is the same in pregnancy as among nonpregnant adults. Both benznidazole and nifurtimox are associated with substantial toxicity in chronic T. cruzi infection. Minimal data are available on potential reproductive toxicity of these drugs, although both drugs have been associated with increased detection of chromosomal aberrations in children being treated for Chagas disease (691--692). Benznidazole crosses the placenta in rats and covalently binds to fetal proteins (693). Because of the toxicity and limited experience with use of these drugs in pregnancy, treatment of acute T. cruzi infection among pregnant women should only be undertaken in consultation with a specialist in this area, and treatment of chronic disease should be considered after completion of the pregnancy. For HIV-1--infected pregnant women with symptomatic reactivation of T. cruzi infection, maximization of the immune response with ART should be the primary approach to therapy (AIII). Perinatal transmission of T. cruzi might occur with acute infection during pregnancy, which has been described rarely, or more often, with reactivation of chronic infection. Perinatal transmission rates among general populations of pregnant women seropositive for antibodies to T. cruzi range from 2%--10% (689,690). The effect of concurrent HIV-1 infection in the mother on risk for perinatal transmission of T. cruzi is not well defined, but the risk for reactivation and transmission might be increased among women with advanced immunosuppression. Infants co-infected with HIV-1 and T. cruzi might be more likely to have symptoms, especially neurologic symptoms (686). References1. Palella FJ Jr, Delaney KM, Moorman AC, et al. Declining morbidity among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med 1998;338:853--60. 2. Tierney C, Lathey JL, Christopherson C, et al. Prognostic value of baseline human immunodeficiency virus type 1 DNA measurement for disease progression in patients receiving nucleoside therapy. J Infect Dis 2003;187:144--8. 3. Press N, Tyndall MW, Wood E, et al. Virologic and immunologic response, clinical progression, and highly active antiretroviral therapy adherence. J Acquir Immune Defic Syndr 2002;31:112--7. 4. Mocroft A, Vella S, Benfield TL, et al. Changing patterns of mortality across Europe in patients infected with HIV-1. Lancet 1998;352: 1725--30. 5. Miller V, Mocroft A, Reiss P, et al. Relations among CD4 lymphocyte count nadir, antiretroviral therapy, and HIV-1 disease progression: results from the EuroSIDA study. Ann Intern Med 1999;130:570--7. 6. Dore GJ, Li Y; McDonald A; Ree H, et al. Impact of highly active antiretroviral therapy on individual AIDS-defining illness incidence and survival in Australia. JAIDS 2002;29:388--95. 7. Currier JS, Williams PL, Grimes, et al. Incidence rates and risk factors for opportunistic infections in a phase III trial comparing indinavir + ZDV + 3TC to ZDV + 3TC. 5th [Abstract]. Conference on Retroviruses and Opportunistic infections, February 1--5, 1998. 8. Autran B, Carcelain G, Li TS, et al. Positive effects of combined antiretroviral therapy on CD4+ T cell homeostasis and function in advanced HIV disease. Science 1997;277:112--6. 9. Connick E, Lederman MM, Kotsin BL et al. Immune reconstitution in the first year of potent antiretroviral therapy and its relationship to virologic response. J Infect Dis 2000;181:358--63. 10. Worrell S, Deayton J, Hayes P, et al. Molecular correlates in AIDS patients following antiretroviral therapy: diversified T-cell receptor repertoires and in vivo control of cytomegalovirus replication. HIV Med 2001;2:11--9. 11. Carr A, Marriott D, Field A, et al. Treatment of HIV-1--associated microsporidiosis and cryptosporidiosis with combination antiretroviral therapy. Lancet 1998;351:256--61. 12. Foudraine NA, Weverling GJ, van Gool T, et al. Improvement of chronic diarrhea in patients with advanced HIV-1 infection during potent antiretroviral therpay. AIDS 1998;12:35--41. 13. Murdaca G, Campelli A, Setti M, et al. Complete remission of AIDS/Kaposi's sarcoma after treatment with a combination of two nucleoside reverse transcriptase inhibitors and one non-nucleoside reverse transcriptase inhibitor [Letter]. AIDS 2002;16:304--5. 14. Tantisiriwat W, Tebas P, Clifford DB, et al. Progressive multifocal leukoencephalopathy in patients with AIDS receiving highly active antiretroviral therapy. Clin Infect Dis 1999;28:1152--4. 15. Egger M, May M, Chene, et al. Prognosis of HIV-1--infected patients starting highly active antiretroviral therapy: a collaborative analysis of prospective studies. Lancet 2002;360:119--29. 16. Phillips P, Kwiatkowski MB, Copland M, et al. Mycobacterial lymphadenitis associated with the initiation of combination ART. J Acquir Immune Defic Syndr Hum Retrovirol 1999;20:122--8. 17. Race EM, Adelson-Mitty J, Kriegel GR, et al. Focal mycobacterial lymphadenitis following initiation of protease-inhibitor therapy in patients with advanced HIV-1 disease. Lancet 1998;351:252--5. 18. Dworkin MS, Fratkin MD. Mycobacterium avium complex lymph node abscess after use of highly active antiretroviral therapy in a patient with AIDS [Letter]. Arch Intern Med 1998;158:1828. 19. Navas E, Martin-Davila P, Moreno L. Paradoxical reactions of tuberculosis in patients with the acquired immunodeficiency syndrome who are treated with highly active antiretroviral therapy. Arch Intern Med 2002;162:97--9. 20. Wislez M, Bergot E, Antoine M, et al. Acute respiratory failure following HAART introduction in patients treated for Pneumocystis carinii pneumonia. Am J Respir Crit Care Med 2001;164:847--51. 21. Lanzafame M, Trevenzoli M, Carreetta G, et al. Mediastinal lymphadenitis due to cryptococcal infection in HIV-positive patients on highly active antiretroviral therapy. Chest 1999;116:848--9. 22. Miralles P, Berenguer J, Lacruz C, et al. Inflammatory reactions in progressive multifocal leukoencephalopathy after highly active antiretroviral therapy. AIDS 2001;15:1900--2. 23. Cinque P, Pierotti C, Vigano MG, et al. The good and evil of HAART in HIV- progressive multifocal leukoencephalopathy. J Neurovirol 2001;7:358--63. 24. Giudici B, Vaz B, Bossolasco S, et al. Highly active antiretroviral therapy and progressive multifocal leukoencephalopathy: effects on cerebrospinal fluid markers of JC virus replication and immune response. Clin Infect Dis 2000;30:95--9. 25. DeSimone JA, Pomerantz RJ, Babinchak TJ. Inflammatory reactions in HIV-1--infected persons after initiation of highly active antiretroviral therapy. Ann Intern Med 2000;133:447--54. 26. Olalla J, Pombo M, Aguadi JM, et al. Paradoxical responses in a cohort of HIV-1--infected patients with mycobacterial disease. Int J Tuberc Lung Dis 2002;6:71--5. 27. Fishman JE, Saraf-Lavi E, Narita M, et al. Pulmonary tuberculosis in AIDS patients: transient chest radiographic worsening after initiation of antiretroviral therapy. AJR Am J Roentgenol 2000;174:43--9. 28. Jacobson MA, Zegans M, Pavan PR, et al. Cytomegalovirus retinitis after initiation of highly active antiretroviral therapy. Lancet 1997;349:1443--5. 29. French MA, Lenzo N, John M, et al. Immune restoration disease after the treatment of immunodeficient HIV-infected patients with highly active antiretroviral therapy. HIV Med 2000;1:107--15. 30. Price P, Mathiot N, Krueger R, et al. Immune dysfunction and immune restoration disease in HIV patients given highly active antiretroviral therapy. J Clin Virol 2001; 22:279--87 31. Fox PA, Barton SE, Francis N, et al. Chronic erosive herpes simplex virus infection of the penis, a possible immune reconstitution disease. HIV Med 1999;1:10--8. 32. Currier JS, Williams PL, Koletar SL, et al. Discontinuation of Mycobacterium avium complex prophylaxis in patients with antiretroviral therapy--induced increases in CD4+ cell count: a randomized, double-blind, placebo-controlled trial. Ann Intern Med 2000;133:493--503. 33. Cinti SK, Kaul DR, Sax PE, et al. Recurrence of Mycobacterium avium infection in patients receiving highly active antiretroviral therapy and antimycobacterial agents. Clin Infect Dis 2000;30:511--4. 34. European Collaborative Study and the Swiss HIV Pregnancy Cohort. Immunological markers in HIV-infected pregnant women. AIDS 1997;11:1859--65. 35. Tuomala RE, Kalish LA, Zorilla C, et al. Changes in total, CD4+, and CD8+ lymphocytes during pregnancy and 1 year postpartum in human immunodeficiency virus--infected women. Obstet Gynecol 1997;89:967--74. 36. Miotti PG, Liomba G, Dallabetta GA, et al. T lymphocyte subsets during and after pregnancy: Analysis in human immunodeficiency virus type 1--infected and --uninfected Malawian mothers. J Infect Dis 1992;165:1116--9. 37. Cruickshank DP, Wigton TR, Hays PM. Maternal physiology in pregnancy. In: Gabbe SG, Neibyl JR, Simpson JL, eds. Obstetrics: Normal and Problem Pregnancies. New York, NY: Churchchill Livingstone, 1996. 38. American College of Obstetricians and Gynecologists Committee. Opinion: guidelines for diagnostic imaging during pregnancy. Number 158, September, 1995. 39. Toppenberg KS, Hill DA, Miller DP. Safety of radiographic imaging during pregnancy. Am Fam Physician 1999;59:1813--8. 40. Adelstein SJ. Administered radionuclides in pregnancy. Teratology 1999;59:236--9. 42. American College of Obstetricians and Gynecologists Practice Bulletin. Antepartum fetal surveillance. October 1999 (No. 9). 43. Pifer LL, Hughes WT, Stagno S, et al. Pneumocystis carinii infection: evidence for high prevalence in normal and immunosuppressed children. Pediatrics 1978;61:35--41. 44. Keely SP, Stringer JR, Baughman RP, et al. Genetic variation among Pneumocystis carinii hominis isolates in recurrent pneumocystosis. J Infect Dis 1995;172:595--8. 45. Helweg-Larsen J, Tsolaki AG, Miller RF, et al. Clusters of Pneumocystis carinii pneumonia: analysis of person-to-person transmission by genotyping. QJM 1998;91:813--20. 46. Phair JP, Munoz A, Detels R, et al. The risk of Pneumocystis carinii pneumonia among men with human immunodeficiency virus type 1. N Engl J Med 1990;322:161--5. 47. Kaplan JE, Hanson DL, Navin TR, et al. Risk factors for primary Pneumocystis carinii pneumonia in human immunodeficiency virus--infected adolescents and adults in the United States: reassessment of indications for chemoprophylaxis. J Infect Dis 1998;178:1126--32. 48. Kaplan JE, Hanson DL, Jones JL, et al. Viral load as an independent risk factor for opportunistic infections in HIV-infected adults and adolescents. AIDS 2001;15:1831--6. 49. Furrer H, Egger M, Opravil M, et al. Discontinuation of primary prophylaxis against Pneumocystis carinii pneumonia in HIV-1--infected adults treated with combination antiretroviral therapy. N Engl J Med 1999;340:1301--6. 50. Lundberg BE, Davidson AJ, Burman WJ. Epidemiology of Pneumocystis carinii pneumonia in an era of effective prophylaxis: the relative contribution of non-adherence and drug failure. AIDS 2000;14:2559--66. 51. Wolff AJ, O'Donnell AE. Pulmonary manifestations of HIV infection in the era of highly active antiretroviral therapy. Chest 2001;120:1888--93. 52. Kovacs JA, Hiemenz JW, Macher AM, et al. Pneumocystis carinii pneumonia: a comparison between patients with the acquired immunodeficiency syndrome and patients with other immunodeficiencies. Ann Intern Med 1984;100:663--71. 53. Selwyn PA, Pumerantz AS, Durante A, et al. Clinical predictors of Pneumocystis carinii pneumonia, bacterial pneumonia and tuberculosis in HIV-infected patients. AIDS 1998;12:885--93. 54. Smith DE, McLuckie A, Wyatt J, Gazzard B, et al. Severe exercise hypoxaemia with normal or near normal X-rays: a feature of Pneumocystis carinii infection. Lancet 1988;2:1049--51. 55. Zaman MK, White DA. Serum lactate dehydrogenase levels and Pneumocystis carinii pneumonia. diagnostic and prognostic significance. Am Rev Respir Dis 1988;137:796--800. 56. Opravil M, Marincek B, Fuchs WA, et al. Shortcomings of chest radiography in detecting Pneumocystis carinii pneumonia. J Acquir Immune Defic Syndr 1994;7:39--45. 57. Baughman RP, Dohn MN, Frame PT. The continuing utility of bronchoalveolar lavage to diagnose opportunistic infection in AIDS patients. Am J Med 1994;97:515--22. 58. Stover DE, Zaman MB, Hajdu SI, Lange M, Gold J, Armstong D. Bronchoalveolar lavage in the diagnosis of diffuse pulmonary infiltrates in the immunosuppressed host. Ann Intern Med 1984;101:1--7. 59. Metersky ML, Colt HG, Olson LK, Shanks TG. AIDS-related spontaneous pneumothorax. Risk factors and treatment. Chest 1995;108:946--51. 60. Sepkowitz KA, Telzak EE, Gold JW, et al. Pneumothorax in AIDS. Ann Intern Med 1991;114:455--9. 61. Gruden JF, Huang L, Turner J, et al. High-resolution CT in the evaluation of clinically suspected Pneumocystis carinii pneumonia in AIDS patients with normal, equivocal, or nonspecific radiographic findings. AJR Am J Roentgenol 1997;169:967--75. 62. Rosso J, Guillon JM, Parrot A, et al. Technetium-99m-DTPA aerosol and gallium-67 scanning in pulmonary complications of human immunodeficiency virus infection. J Nucl Med 1992;33:81--7. 63. Kovacs JA, Ng VL, Masur H, et al. Diagnosis of Pneumocystis carinii pneumonia: improved detection in sputum with use of monoclonal antibodies. N Engl J Med 1988;318:589--93. 64. Roger PM, Vandenbos F, Pugliese P, et al. Persistence of Pneumocystis carinii after effective treatment of P. carinii pneumonia is not related to relapse or survival among patients infected with human immunodeficiency virus. Clin Infect Dis 1998;26:509--10. 65. Larsen HH, Masur H, Kovacs JA, et al. Development and evaluation of a quantitative, touch-down, real-time PCR assay for diagnosing Pneumocystis carinii pneumonia. J Clin Microbiol 2002;40:490--4. 66. Torres J, Goldman M, Wheat LJ, et al. Diagnosis of Pneumocystis carinii pneumonia in human immunodeficiency virus--infected patients with polymerase chain reaction: a blinded comparison to standard methods. Clin Infect Dis 2000;30:141--45. 67. Hughes W, Leoung G, Kramer F, et al. Comparison of atovaquone (566C80) with trimethoprim-sulfamethoxazole to treat Pneumocystis carinii pneumonia in patients with AIDS. N Engl J Med 1993;328:1521--7. 68. Safrin S, Finkelstein DM, Feinberg J, et al. Comparison of three regimens for treatment of mild to moderate Pneumocystis carinii pneumonia in patients with AIDS. A double-blind, randomized trial of oral trimethoprim-sulfamethoxazole, dapsone-trimethoprim, and clindamycin-primaquine. Ann Intern Med 1996;12:792--802. 69. Safrin S, Lee BL, Sande MA. Adjunctive folinic acid with trimethoprim-sulfamethoxazole for Pneumocystis carinii pneumonia in AIDS patients is associated with an increased risk of therapeutic failure and death. J Infect Dis 1994;170:912--7. 70. Navin TR, Beard CB, Huang L, et al. Effect of mutations in Pneumocystis carinii dihydropteroate synthase gene on outcome of P carinii pneumonia in patients with HIV-1: a prospective study. Lancet 2001;358:545--9. 71. Nielsen TL, Eeftinck Schattenkerk JK, Jensen BN, et al. Adjunctive corticosteroid therapy for Pneumocystis carinii pneumonia in AIDS: a randomized European multicenter open label study. J Acquir Immune Defic Syndr 1992;5:726--31. 72. Bozzette SA, Sattler FR, Chiu J, et al. A controlled trial of early adjunctive treatment with corticosteroids for Pneumocystis carinii pneumonia in the acquired immunodeficiency syndrome. N Engl J Med 1990;323:1451--7. 73. National Institutes of Health--University of California Expert Panel for Corticosteroids as Adjunctive Therapy for Pneumocystis Pneumonia. Consensus statement on the use of corticosteroids as adjunctive therapy for Pneumocystis pneumonia in the acquired immunodeficiency syndrome. N Engl J Med 1990;323:1500--4. 74. Montaner JS, Lawson LM, Levitt N, Belzberg A, Schechter MT, Ruedy J. Corticosteroids prevent early deterioration in patients with moderately severe Pneumocystis carinii pneumonia and the acquired immunodeficiency syndrome (AIDS). Ann Intern Med 1990;113:14--20. 75. Gallant JE, Chaisson RE, Moore RD. The effect of adjunctive corticosteroids for the treatment of Pneumocystis carinii pneumonia on mortality and subsequent complications. Chest 1998;114:1258--63. 76. Medina I, Mills J, Leoung, et al. Oral therapy for Pneumocystis carinii pneumonia in the acquired immunodeficiency syndrome. A controlled trial of trimethoprim-sulfamethoxazole versus trimethoprim-dapsone. N Engl J Med 1990;323:776--82. 77. Black JR, Feinberg J, Murphy RL, et al. Clindamycin and primaquine therapy for mild-to-moderate episodes of Pneumocystis carinii pneumonia in patients with AIDS. Clin Infect Dis 1994;18:905--13. 78. Toma E, Thorne A, Singer J, et al. Clindamycin with primaquine vs. trimethoprim-sulfamethoxazole therapy for mild and moderately severe Pneumocystis carinii pneumonia in patients with AIDS: a multicenter, double-blind, randomized trial. Clin Infect Dis 1998;27: 524--30. 79. Smego RA Jr., Nagar S, Maloba B, Propara M. A meta-analysis of salvage therapy for Pneumocystis carinii pneumonia. Arch Intern Med 2001;161:1529--33. 80. Wharton JM, Coleman DL, Wofsy CB, et al. Trimethoprim-sulfamethoxazole or pentamidine for Pneumocystis carinii pneumonia in the acquired immunodeficiency syndrome. A prospective randomized trial. Ann Intern Med 1986;105:37--44. 81. Klein NC, Duncanson FP, Lenox TH, et al. Trimethoprim-sulfamethoxazole versus pentamidine for Pneumocystis carinii pneumonia in AIDS patients: results of a large prospective randomized treatment trial. AIDS 1992;6:301--5. 82. Conte JE Jr., Chernoff D, Feigal DW Jr., Joseph P, McDonald C, Golden JA. Intravenous or inhaled pentamidine for treating Pneumocystis carinii pneumonia in AIDS. A randomized trial. Ann Intern Med 1990;113:203--9. 83. Dohn MN, Weinberg WG, Torres RA, et al. Oral atovaquone compared with intravenous pentamidine for Pneumocystis carinii pneumonia in patients with AIDS. Ann Intern Med 1994;121:174--80. 84. Sattler FR, Frame P, Davis R, et al. Trimetrexate with leucovorin versus trimethoprim-sulfamethoxazole for moderate to severe episodes of Pneumocystis carinii pneumonia in patients with AIDS. J Infect Dis 1994;170:165--72. 85. Soo Hoo GW, Mohsenifar Z, Meyer RD. Inhaled or intravenous pentamidine therapy for Pneumocystis carinii pneumonia in AIDS. A randomized trial. Ann Intern Med 1990;113:195--202. 86. Montgomery AB, Feigal DW Jr., Sattler F, et al. Pentamidine aerosol versus trimethoprim-sulfamethoxazole for Pneumocystis carinii in acquired immune deficiency syndrome. Am J Respir Crit Care Med 1995;151:1068--74. 87. Randall CJ, Yarnold PR, Schwartz DN, Weinstein RA, Bennett CL. Improvements in outcomes of acute respiratory failure for patients with human immunodeficiency virus--related Pneumocystis carinii pneumonia. Am J Respir Crit Care Med 2000;162(2 Pt 1):393--8. 88. Dworkin MS, Hanson DL, Navin TR. Survival of patients with AIDS, after diagnosis of Pneumocystis carinii pneumonia, in the United States. J Infect Dis 2001;183:1409--12. 89. Morris A, Wachter RM, Luce J, Turner J, Huang L. Improved survival with highly active antiretroviral therapy in HIV-infected patients with severe Pneumocystis carinii pneumonia. AIDS 2003;17:73--80. 90. Wislez M, Bergot E, Antoine M, et al. Acute respiratory failure following HAART introduction in patients treated for Pneumocystis carinii pneumonia. Am J Respir Crit Care Med 2001;164:847--51. 91. Hughes WT, LaFon SW, Scott JD, Masur H. Adverse events associated with trimethoprim-sulfamethoxazole and atovaquone during the treatment of AIDS-related Pneumocystis carinii pneumonia. J Infect Dis 1995;171:1295--301. 92. Gordin FM, Simon GL, Wofsy CB, Mills J. Adverse reactions to trimethoprim-sulfamethoxazole in patients with the acquired immunodeficiency syndrome. Ann Intern Med 1984;100:495--9. 93. Eeftinck Schattenkerk JK, Lange JM, van Steenwijk RP, Danner SA. Can the course of high dose cotrimoxazole for Pneumocystis carinii pneumonia in AIDS be shorter? A possible solution to the problem of cotrimoxazole toxicity. J Intern Med 1990;227:359--62. 94. Kaplan J, Masur H, Holmes K. Guidelines for preventing opportunistic infections among HIV-infected persons---2002. Recommendations of the U.S. Public Health Service and the Infectious Diseases Society of America. MMWR 2002;51(No. RR-8). 95. Ledergerber B, Mocroft A, Reiss P, et al. Discontinuation of secondary prophylaxis against Pneumocystis carinii pneumonia in patients with HIV infection who have a response to antiretroviral therapy. N Engl J Med 2001;344:168--74. 96. Mussini C, Pezzotti P, Antinori A, et al. Discontinuation of secondary prophylaxis for Pneumocystis carinii pneumonia in human immuno--deficiency virus--infected patients. Clin Infect Dis 2003;36: 645--51. 97. Furrer H, Egger M, Opravil M, et al. Discontinuation of primary prophylaxis against Pneumocystis carinii pneumonia in HIV-1--infected adults treated with combination antiretroviral therapy. N Engl J Med 1999;340:1301--6. 98. Connelly RT, Lourwood DL. Pneumocystis carinii pneumonia prophylaxis during pregnancy. Pharmacotherapy 1994;14:424--9. 99. Andersen DH, Blanc WA, Crozier DN, Silverman WA. A difference in mortality rate and incidence of kernicterus among premature infants allotted to two prophylactic antibacterial regimens. Pediatrics 1956;18:614--25. 100. Harstad TW, Little BB, Bawdon RE, Kroll K, Rod D, Gilstrap LC 3rd. Embryofetal effects of pentamidine isothionate administered to pregnant Sprague-Dawley rats. Am J Obstet Gynecol 1990:163:912--9. 101. Lloyd ME, Carr M, McElhatton P, Hall GM, Hughes RA. The effects of methotrexate on pregnancy, fertility and lactation. QJM 1999;92:551--63. 102. Albino JA, Shapiro JM. Respiratory failure in pregnancy due to Pneumocystis carinii: report of a successful outcome. Obstet Gynecol 1994;83(5 Pt 2):823--4. 103. Madinger NE, Greenspoon JS, Ellrodt AG. Pneumonia during pregnancy: has modern technology improved maternal and fetal outcome? Am J Obstet Gynecol 1989;161:657--62. 104. Koonin LM, Ellerbrock TV, Atrash HK, et al. Pregnancy-associated deaths due to AIDS in the United States. JAMA 1989;261:1306--9. 105. Benedetti TJ, Valle R, Ledger WJ. Antepartum pneumonia in pregnancy. Am J Obstet Gynecol 1982;144:413--7. 106. Luft BJ, Conley F, Remington JS, et al. Outbreak of central-nervous-system toxoplasmosis in western Europe and North America. Lancet 1983;1:781--3. 107. Luft BJ, Brooks RG, Conley FK, McCabe RE, Remington JS. Toxoplasmic encephalitis in patients with acquired immune deficiency syndrome. JAMA 1984;252:913--7. 108. Wong B, Gold JW, Brown AE, et al. Central-nervous-system toxoplasmosis in homosexual men and parenteral drug abusers. Ann Intern Med 1984;100:36--42. 109. Israelski DM, Chmiel JS, Poggensee L, Phair JP, Remington JS. Prevalence of Toxoplasma infection in a cohort of homosexual men at risk of AIDS and toxoplasmic encephalitis. J Acquir Immune Defic Syndr 1993;6:414--8. 110. Mathews WC, Fullerton SC. Use of a clinical laboratory database to estimate Toxoplasma seroprevalence among human immunodeficiency virus--infected patients. Overcoming bias in secondary analysis of clinical records. Arch Pathol Lab Med 1994;118:807--10. 111. Duval X, Leport C. Toxoplasmosis in AIDS. Current Treatment Options in Infectious Diseases 2001;3:113--28. 112. Abgrall S, Rabaud C, Costagliola D; Clinical Epidemiology Group of the French Hospital Database on HIV. Incidence and risk factors for toxoplasmic encephalitis in human immunodeficiency virus--infected patients before and during the highly active antiretroviral therapy era. Clin Infect Dis 2001;33:1747--55. 113. Leport C, Chene G, Morlat P, et al. Pyrimethamine for primary prophylaxis of toxoplasmic encephalitis in patients with human immunodeficiency virus infection: a double-blind, randomized trial. J Infect Dis 1996;173:91--7. 114. Kupfer MC, Zee CS, Colletti PM, Boswell WD, Rhodes R. MRI evaluation of AIDS-related encephalopathy: toxoplasmosis vs. lymphoma. Magn Reson Imaging 1990;8:51--7. 115. Pierce MA, Johnson MD, Maciunas RJ, et al. Evaluating contrast-enhancing brain lesions in patients with AIDS by using positron emission tomography. Ann Intern Med 1995;123:594--8. 116. Ruiz A, Ganz WI, Post MJ, et al. Use of thallium-201 brain SPECT to differentiate cerebral lymphoma from toxoplasma encephalitis in AIDS patients. Am J Neuroradiol 1994;15:1885--94. 117. Derouin F, Leport C, Pueyo S, et al. Predictive value of Toxoplasma gondii antibody titres on the occurrence of toxoplasmic encephalitis in HIV-infected patients. AIDS 1996;10:1521--7. 118. Conley FK, Jenkins KA, Remington JS. Toxoplasma gondii infection of the central nervous system. Use of the peroxidase-antiperoxidase method to demonstrate toxoplasma in formalin fixed, paraffin embedded tissue sections. Hum Pathol 1981;12:690--8. 119. Novati R, Castagna A, Morsica G, et al. Polymerase chain reaction for Toxoplasma gondii DNA in the cerebrospinal fluid of AIDS patients with focal brain lesions. AIDS 1994;8:1691--4. 120. Cinque P, Scarpellini P, Vago L, Linde A, Lazzarin A. Diagnosis of central nervous system complications in HIV-infected patients: cerebrospinal fluid analysis by the polymerase chain reaction. AIDS 1997;11:1--17. 121. Skiest DJ. Focal neurological disease in patients with acquired immunodeficiency syndrome. Clin Infect Dis 2002;34:103--15. 122. Katlama C, De Wit S, O'Doherty E, Van Glabeke M, Clumeck N. Pyrimethamine-clindamycin vs. pyrimethamine-sulfadiazine as acute and long-term therapy for toxoplasmic encephalitis in patients with AIDS. Clin Infect Dis 1996;22:268--75. 123. Dannemann BR, McCutchan JA, Israelski DM, et al. Treatment of toxoplasmic encephalitis in patients with AIDS: a randomized trial comparing pyrimethamine plus clindamycin to pyrimethamine plus sulfadiazine. Ann Intern Med 1992;116:33--43. 124. Leport C, Raffi F, Matheron S, et al. Treatment of central nervous system toxoplasmosis with pyrimethamine/sulfadiazine combination in 35 patients with the acquired immunodeficiency syndrome. Efficacy of long-term continuous therapy. Am J Med 1988;84:94--100. 125. Luft BJ, Hafner R, Korzun AH, et al. Toxoplasmic encephalitis in patients with the acquired immunodeficiency syndrome. N Engl J Med 1993;329:995--1000. 126. Leport C, Meulemans A, Robine D, Dameron G, Vilde JL. Levels of pyrimethamine in serum and penetration into brain tissue in humans [Letter]. AIDS 1992;6:1040--1. 127. Van Delden C, Hirschel B. Folinic acid supplements to pyrimethamine-sulfadiazine for Toxoplasma encephalitis are associated with better outcome [Letter]. J Infect Dis 1996;173:1294--5. 128. Frenkel JK, Hitchings GH. Relative reversal by vitamins (p-aminobenzoic,folic, and folinic acids) of the effects of sulfadiazine and pyrimethamine on Toxoplasma, mouse and man. Antibiot Chemothe 1957;7:630--8. 129. Torre D, Casari S, Speranza F, et al. Randomized trial of trimethoprim-sulfamethoxazole versus pyrimethamine-sulfadiazine for therapy of toxoplasmic encephalitis in patients with AIDS. Antimicrob Agents Chemother 1998;42:1346--9. 130. Chirgwin K, Hafner R, Leport C, et al. Randomized phase II trial of atovaquone with pyrimethamine or sulfadiazine for treatment of toxoplasmic encephalitis in patients with acquired immunodeficiency syndrome. Clin Infect Dis 2002;34:1243--50. 131. Kovacs JA. Efficacy of atovaquone in treatment of toxoplasmosis in patients with AIDS. Lancet 1992;340:637--8. 132. Torres RA, Weinberg W, Stansell J, et al. Atovaquone for salvage treatment and suppression of toxoplasmic encephalitis in patients with AIDS. Clin Infect Dis 1997;24:422--9. 133. Katlama C, Mouthon B, Gourdon D, Lapierre D, Rousseau F. Atovaquone as long-term suppressive therapy for toxoplasmic encephalitis in patients with AIDS and multiple drug intolerance. AIDS 1996;10:1107--12. 134. Saba J, Morlat P, Raffi F, et al. Pyrimethamine plus azithromycin for treatment of acute toxoplasmic encephalitis in patients with AIDS. Eur J Clin Microbiol Infect Dis 1993;12:853--6. 135. Jacobson JM, Hafner R, Remington J, et al. Dose-escalation, phase I/II study of azithromycin and pyrimethamine for the treatment of toxoplasmic encephalitis in AIDS. AIDS 2001;15:583--9. 136. Fernandez-Martin J, Leport C, Morlat P, Meyohas MC, Chauvin JP, Vilde JL. Pyrimethamine-clarithromycin combination for therapy of acute Toxoplasma encephalitis in patients with AIDS. Antimicrob Agents Chemother 1991;35:2049--52. 137. Dhiver C, Milandre C, Poizot-Martin I, Drogoul MP, Gastaut JL, Gastaut JA. 5-Fluoro-uracil-clindamycin for treatment of cerebral toxoplasmosis. AIDS 1993;7:143--4. 138. Derouin F, Piketty C, Chastang C, Chau F, Rouveix B, Pocidalo JJ. Anti-Toxoplasma effects of dapsone alone and combined with pyrimethamine. Antimicrob Agents Chemother 1991;35:252--5. 139. Lacassin F, Schaffo D, Perronne C, Longuet P, Leport C, Vilde JL. Clarithromycin-minocycline combination as salvage therapy for toxoplasmosis in patients infected with human immunodeficiency virus [Letter]. Antimicrob Agents Chemother 1995;39:276--7. 140. Hagberg L, Palmertz B, Lindberg J. Doxycycline and pyrimethamine for toxoplasmic encephalitis. Scand J Infect Dis 1993;25:157--60. 141. Podzamczer D, Miro JM, Bolao F, et al. Twice-weekly maintenance therapy with sulfadiazine-pyrimethamine to prevent recurrent toxoplasmic encephalitis in patients with AIDS. Ann Intern Med 1995;123: 175--80. 142. Leport C, Tournerie C, Raguin G, et al. Long-term follow-up of patients with AIDS on maintenance therapy for toxoplasmosis. Eur J Clin Microbiol Infect Dis 1991;10:191--3. 143. Podzamczer D, Miro JM, Ferrer E, et al. Thrice-weekly sulfadiazine-pyrimethamine for maintenance therapy of toxoplasmic encephalitis in HIV-infected patients. Eur J Clin Microbiol Infect Dis 2000;19:89--95. 144. Kirk O, Reiss P, Uberti-Foppa C, et al. Safe interruption of maintenance therapy against previous infection with four common HIV-associated opportunistic pathogens during potent antiretroviral therapy. Ann Intern Med 2002;137:239--50. 145. Zeller V, Truffot C, Agher R, et al. Discontinuation of secondary prophylaxis against disseminated Mycobacterium avium complex infection and toxoplasmic encephalitis. Clin Infect Dis 2002;34:662--7. 146. Mitchell CD, Erlich SS, Mastrucci MT, Hutto SC, Parks WP, Scott GB. Congenital toxoplasmosis occurring in infants perinatally infected with human immunodeficiency virus 1. Pediatr Infect Dis J 1990;9:512--8. 147. Dunn D, Newell M-L, Gilbert R, Mok J, Petersen E, Peckham C. Low incidence of congenital toxoplasmosis in children born to women infected with human immunodeficiency virus. European Journal of Obstetrics & Gynecology and Reproductive Biology 1996;68:93--6. 148. Wong S-Y, Remington JS. Toxoplasmosis in pregnancy. Clin Infect Dis 1994;18:853--61. 149. Minkoff H, Remington JS, Holman S, Ramirez R, Goodwin S, Landesman S. Vertical transmission of Toxoplasma by human immunodeficiency virus--infected women. Am J Obstet Gynecol 1997;176: 555--9. 150. Flanigan TP, Whalen C, Toerner J, et al. Cryptosporidium infection and CD4 counts. Ann Intern Med 1992;116:840--2 151. Goodgame RW: Understanding intestinal spore-forming protozoa: cryptosporidia, microsporidia, Isospora, and cyclospora. Ann Intern Med 1996;124:429--41. 152. Ducreux M, Buffet C, Lamy P, et al. Diagnosis and prognosis of AIDS-related cholangitis. AIDS 1995;9:875--80. 153. Weber R, Bryan RT, Bishop HS, Wahlquist SP, Sullivan JJ, Juranek DD. Threshold of detection of Cryptosporidium oocysts in human stool specimens: evidence for low sensitivity of current diagnostic methods. J Clin Microbiol 1991;29:1323--7. 154. Miao YM, Awad-El-Kariem FM, Franzen C, et al. Eradication of cryptosporidia and microsporidia following successful antiretroviral therapy. J Acquir Immune Defic Syndr 2000;25:124--9. 155. Carr A, Marriott D, Field A, Vasak E, Cooper DA. Treatment of HIV-1--associated microsporidiosis and cryptosporidiosis with combination antiretroviral therapy. Lancet 1998;351:256--61. 156. Tzipori S, Rand W, Griffiths J, Widmer G, Crabb J. Evaluation of an animal model system for cryptosporidiosis: therapeutic efficacy of paromomycin and hyperimmune bovine colostrum-immunoglobulin. Clin Diagn Lab Immunol 1994;1:450--63. 157. Hewitt RG, Yiannoutsos CT, Higgs ES, et al. Paromomycin: no more effective than placebo for treatment of cryptosporidiosis in patients with advanced human immunodeficiency virus infection. Clin Infect Dis 2000;31:1084--92. 158. White AC Jr, Chappell CL, Hayat CS, Kimball KT, Flanigan TP, Goodgame RW. Paromomycin for cryptosporidiosis in AIDS: a prospective, double-blind trial. J Infect Dis 1995;170:419--24. 159. Smith NH, Cron S, Valdez LM, Chappell CL, White AC Jr. Combination drug therapy for cryptosporidiosis in AIDS. J Infect Dis 1998;178:900--3. 160. Rossignol JF, Ayoub A, Ayers MS. Treatment of diarrhea caused by Cryptosporidium parvum: a prospective randomized, double-blind, placebo-controlled study of nitazoxanide. J Infect Dis 2001;184:103--6. 161. Rossignol JF, Hidalgo H, Feregrino M, et al. A double-`blind' placebo-controlled study of nitazoxanide in the treatment of cryptosporidial diarrhoea in AIDS patients in Mexico. Trans R Soc Trop Med Hyg 1998;92:663--6. 162. Simon DM, Cello JP, Valenzuela J, et al. Multicenter trial of octreotide in patients with refractory acquired immunodeficiency syndrome-associated diarrhea. Gastroenterology 1995;108:1753--60. 163. Wittner M, Weiss LM, eds. The microsporidia and microsporidiosis. Washington, DC: ASM Press, 1999. 164. Mathis, A. Microsporidia: emerging advances in understanding the basic biology of these unique organisms. Int J Parasitol 2000;30: 795--804. 165. Deplazes P, Mathis A, Weber R. Epidemiology and zoonotic aspects of microsporidia of mammals and birds. Contrib Microbiol 2000;6:236--60. 166. Kotler DP, Orenstein JM. Clinical syndromes associated with microsporidiosis. Adv Parasitol 1998;40:321--49. 167. Weber R, Bryan RT, Owen RL, Wilcox CM, Gorelkin L, Visvesvara GS. Improved light-microscopical detection of microsporidia spores in stool and duodenal aspirates. N Engl J Med 1992;326:161--6. 168. Beauvais B, Sarfati C, Molina JM, Lesourd A, Lariviere M, Derouin F. Comparative evaluation of five diagnostic methods for demonstrating microsporidia in stool and intestinal biopsy specimens. Ann Trop Med Parasitol 1993;87:99--102. 169. Weiss LM, Vossbrinck CR. Microsporidiosis: molecular and diagnostic aspects. Adv Parasitol 1998;40:351--95. 170. Maggi P, Larocca AM, Quarto M, et al. Effect of antiretroviral therapy on cryptosporidiosis and microsporidiosis in patients infected with human immunodeficiency virus type 1. Eur J Clin Microbiol Infect Dis 2000;19:213--7. 171. Goguel J, Katlama C, Sarfati C, Maslo C, Leport C, Molina J-M. Remission of AIDS-associated intestinal microsporidiosis with highly active antiretroviral therapy. AIDS 1997;11:1658--9. 172. Conteas CN, Berlin OG, Speck CE, Pandhumas SS, Lariviere MJ, Fu C. Modification of the clinical course of intestinal microsporidiosis in acquired immunodeficiency syndrome patients by immune status and anti--human immunodeficiency virus therapy. Am J Trop Med Hyg 1998;58:555--8. 173. Molina JM, Goguel J, Sarfati C, et al. Trial of oral fumagillin for the treatment of intestinal microsporidiosis in patients with HIV infection [Letter]. AIDS 2000;14:1341--48. 174. Molina JM, Tourneur M, Sarfati C, et al. Fumagillin treatment of intestinal microsporidiosis. N Engl J Med 2002;346:1963--9. 175. Bicart-See A, Massip P, Linas MD, Datry A. Successful treatment with nitazoxanide of Enterocytozoon bieneusi microsporidiosis in a patient with AIDS. Antimicrob Agents Chemother 2000;44:167--8. 176. Didier ES. Effects of albendazole, fumagillin, and TNP-470 on microsporidial replication in vitro. Antimicrob Agents Chemother 1997;41:1541--6. 177. Katiyar SK, Edlind TD. In vitro susceptibilities of the AIDS-associated microsporidian Encephalitozoon intestinalis to albendazole, its sulfoxide metabolite, and 12 additional benzimidazole derivatives. Antibicrob Agents Chemother 1997;41:2729--32. 178. Molina JM, Chastang C, Goguel J, et al. Albendazole for treatment and prophylaxis of microsporidiosis due to Encephalitozoon intestinalis in patients with AIDS: a randomized double-blind controlled trial. J Infect Dis 1998;177:1373--7. 179. Gritz, DC, Holsclaw DS, Neger RE, Whitcher JP Jr, Margolis TP. Ocular and sinus microsporidial infection cured with systemic albendazole. Am J Ophthalmol 1997;124:241--3. 180. Diesenhouse MC, Wilson LA, Corrent GF, Visvesvara GS, Grossniklaus HE, Bryan RT. Treatment of microsporidial keratoconjunctivitis with topical fumagillin. Am J Ophthalmol 1993;115:293--8. 181. Dieterich DT, Lew EA, Kotler DP, Poles MA, Orenstein JM. Treatment with albendazole for intestinal disease due to Enterocytozoon bieneusi in patients with AIDS. J Infect Dis 1994;169:178--83. 182. Dye C, Scheele S, Dolin P, Pathania V, Raviglione MC. Global burden of tuberculosis: estimated incidence, prevalence, and mortality by country. JAMA 1999;282:677--86. 183. Corbett EL, Watt CJ, Walker N, et al. The growing burden of tuberculosis: global trends and interactions with the HIV epidemic. Arch Intern Med 2003;163:1009--21. 184. CDC. World TB day---March 24, 2002. MMWR 2002;51:229. 185. CDC. Treatment of tuberculosis. MMWR 2003;52(No. RR-11). 186. Girardi E, Raviglione MC, Antonucci G, Godfrey-Faussett P, Ippolito G. Impact of the HIV epidemic on the spread of other diseases: the case of tuberculosis. AIDS 2000;14(Suppl 3):47--56. 187. Whalen C, Horsburgh CR, Hom D, Lahart C, Simberkoff M, Ellner J. Accelerated course of human immunodeficiency virus infection after tuberculosis. Am J Respir Crit Care Med 1995;151:129--35. 188. Batungwanayo J, Taelman H, Dhote R, Bogaerts J, Allen S, Van de Perre P. Pulmonary tuberculosis in Kigali, Rwanda. Impact of human immunodeficiency virus infection on clinical and radiographic presentation. Am Rev Respir Dis 1992;146:53--6. 189. Jones BE, Young SM, Antoniskis D, Davidson PT, Kramer F, Barnes PF. Relationship of the manifestations of tuberculosis to CD4 cell counts in patients with human immunodeficiency virus infection. Am Rev Respir Dis 1993;148:1292--7. 190. Perlman DC, El-Sadr WM, Nelson ET, et al. Variation of chest radiographic patterns in pulmonary tuberculosis by degree of human immunodeficiency virus-related immunosuppression. Clin Infect Dis 1997;25:242--6. 191. Smith RL, Yew K, Berkowitz KA, Aranda CP. Factors affecting the yield of acid-fast sputum smears in patients with HIV and tuberculosis. Chest 1994;106:684--6. 192. American Thoracic Society/CDC. Targeted tuberculin testing and treatment of latent tuberculosis infection. MMWR 2000;49 (No. RR-6):1--51. 193. Hsieh SM, Hung CC, Chen MY, Hsueh PR, Chang SC, Luh KT. The role of tissue studies in facilitating early initiation of antimycobacterial treatment in AIDS patients with disseminated mycobacterial disease. Int J Tuberc Lung Dis 1999;3:521--7. 194. Relkin F, Aranda CP, Garay DM, Smith R, Berkowitz KA, Rom WN. Pleural tuberculosis and HIV infection. Chest 1994;105:1338--41. 195. Shriner KA, Mathisen GE, Goetz MB. Comparison of mycobacterial lymphadenitis among persons infected with human immunodeficiency virus and seronegative controls. Clin Infect Dis 1992;15:601--5. 196. Perriens JH, St. Louis ME, Mukadi YB, et al. Pulmonary tuberculosis in HIV-infected patients in Zaire. A controlled trial of treatment for either 6 or 12 months. N Engl J Med 1995;332:779--84. 197. Vernon A, Burman W, Benator D, Khan A, Bozeman L. Acquired rifamycin monoresistance in patients with HIV-related tuberculosis treated with once-weekly rifapentine and isoniazid. Tuberculosis Trials Consortium. Lancet 1999;353:1843--7. 200. Dean GL, Edwards SG, Ives NJ, et al. Treatment of tuberculosis in HIV-infected persons in the era of highly active antiretroviral therapy. AIDS 2002;16:75--83. 201. Veldkamp AI, Hoetelmans MW, Beijnen JH, Mulder JW, Meenhorst PL. Ritonavir enables combined therapy with rifampin and saquinavir. Clin Infect Dis 1999;29:1586. 202. Narita M, Ashkin D, Hollender ES, Pitchenik AE. Paradoxical worsening of tuberculosis following antiretroviral therapy in patients with AIDS. Am J Respir Crit Care Med 1998;158:157--61. 203. Wendel KA, Alwood KS, Gachuhi R, Chaisson RE, Bishai WR, Sterling TR. Paradoxical worsening of tuberculosis in HIV-infected persons Chest;120:193--7. 204. Chien JW, Johnson JW. Paradoxical reactions in HIV and pulmonary TB. Chest 1998;114:933--6. 205. DeSimone JA, Pomerantz RJ, Babinchak TJ. Inflammatory reactions in HIV-1--infected persons after initiation of highly active antiretroviral therapy. Ann Intern Med 2000;133:447--54. 206. Navas E, Martín-Dávila P, Moreno L, et al. Paradoxical reactions of tuberculosis in patients with the acquired immunodeficiency syndrome who are treated with highly active antiretroviral therapy. Arch Intern Med 2002;162:97--9. 207. Mofenson LM, Rodriguez EM, Hershow R, et al. Mycobacterium tuberculosis infection in pregnant and nonpregnant women infected with HIV in the Women and Infants Transmission Study. Arch Intern Med 1995;155:1066--72. 208. Eriksen NL, Helfgott AW. Cutaneous anergy in pregnant and nonpregnant women with human immunodeficiency virus. Infect Dis Obstet Gynecol 1998;6:13--7. 209. Jana N, Vasishta K, Jindal SK, Khunnu B, Ghosh K. Perinatal outcome in pregnancies complicated by pulmonary tuberculosis. Int J Gynaecol Obstet 1994;44:119--24. 210. Jana N, Vasishta K, Saha AC, Ghosh K. Obstetrical outcomes among women with extrapulmonary tuberculosis. N Engl J Med 1999;341:645--9. 211. Brost BC, Newman RB. The maternal and fetal effects of tuberculosis therapy. Obstet Gynecol Clin North Am 1997;24:659--73. 212. Bothamley G. Drug treatment for tuberculosis during pregnancy: safety considerations. Drug Saf 2001;24:553--65. 213. Czeizel AE, Rockenbauer M, Olsen J, Sorensen HT. A population-based case-control study of the safety of oral antituberculosis drug treatment during pregnancy. Int J Tuberc Lung Dis 2001;5:564--8. 214. Franks AL, Binkin NJ, Snider DE, Rokaw WM, Becker S. Isoniazid hepatitis among pregnant and nonpregnant Hispanic patients. Public Health Rep 1989;104:151--5. 215. World Health Organization. Treatment of tuberculosis: guidelines for national programs. 2nd edition. WHO/TB/97.220. Geneva, Switzerland: World Health Organization, 1997. 216. Enarson DA, Rieder HL, Arnodottir T, Trébucq A. Management of tuberculosis: a guide for low income countries. 4th edition. Paris, France: International Union Against Tuberculosis and Lung Disease, 1996. 217. Dluzniewski A, Gastol-Lewinska L. The search for teratogenic activity of some tuberculostatic drugs. Diss Pharm Pharmacol 1971;23: 383--92. 218. Lowe CR. Congenital defects among children born to women under supervision or treatment for pulmonary tuberculosis. Br J Prev Soc Med 1964;18:14--6. 219. Kemper CA, Havlir D, Bartok AE, Kane C, Camp B, Lane N, Deresinski SC. Transient bacteremia due to Mycobacterium avium complex in patients with AIDS. J Infect Dis 1994;170:488--93. 220. Gordin FM, Cohn DL, Sullam PM, Schoenfelder JR, Wynee BA, Horsburgh CR Jr: Early manifestations of disseminated Mycobacterium avium complex disease: a prospective evaluation. J Infect Dis 1997;176:126--32. 221. Inderlied CB, Kemper CA, Bermudez LE. The Mycobacterium avium complex. Clin Microbiol Rev 1993;6:266--310. 222. Benson CA, Ellner JJ. Mycobacterium avium complex infection and AIDS: advances in theory and practice. Clin Infect Dis 1993;17:7--20. 223. Havlik JA Jr, Horsburgh CR Jr, Metchock B, Williams PP, Fann SA, Thompson SE 3rd. Disseminated Mycobacterium avium complex infection: clinical identification and epidemiologic trends. J Infect Dis 1992;165:577--80. 224. Benson CA: Disease due to the Mycobacterium avium complex in patients with AIDS: epidemiology and clinical syndrome. Clin Infect Dis 1994;18(Suppl):218--22. 225. Packer SJ, Cesario T, Williams JH Jr. Mycobacterium avium complex infection presenting as endobronchial lesions in immunosuppressed patients. Ann Intern Med 1988;109:389--93. 226. Barbaro DJ, Orcutt VL, Coldiron BM. Mycobacterium avium--Mycobacterium intracellulare infection limited to the skin and lymph nodes in patients with AIDS. Rev Infect Dis 1989;11:625--8. 227. Hellyer TJ, Brown IN, Taylor MB, Allen BW, Easmon CS. Gastrointestinal involvement in Mycobacterium avium-intracellulare infection of patients with HIV. J Infect 1993;26:55--66. 228. Owen RL, Roth RI, St. Hilaire RJ, Keren DF. Pseudo Whipple's disease---intestinal infection with Mycobacterium avium intracellulare (M. avium) in acquired immune deficiency syndrome (AIDS). Gastroenterology 1983;84:1267. 229. Torriani FJ, McCutchan JA, Bozzette SA, Grafe MR, Havlir DV. Autopsy findings in AIDS patients with Mycobacterium avium complex bacteremia. J Infect Dis 1994;170:1601--5. 230. Phillips P, Kwiatkowski MB, Copland M, Craib K, Montaner J. Mycobacterial lymphadenitis associated with the initiation of combination antiretroviral therapy. J Acquir Immune Defic Syndr Hum Retrovirol 1999;20:122--8. 231. Race EM, Adelson, Mitty J, et al. Focal mycobacterial lymphadenitis following initiation of protease-inhibitor therapy in patients with advanced HIV-1 disease. Lancet 1998;351:252--5. 232. Cabie A, Abel S, Brebion A, Desbois N, Sobesky G. Mycobacterial lymphadenitis after initiation of highly active antiretroviral therapy. Eur J Clin Microbiol Infect Dis 1998;17:812--3. 233. Nightingale SD, Byrd LT, Southern PM, Jockusch JD, Cal SX, Wynne BA. Incidence of Mycobacterium avium--intracellulare complex bacteremia in human immunodeficiency virus--positive patients. J Infect Dis 1992;165:1082--5. 234. Chaisson RE, Moore RD, Richman DD, Keruly H, Creagh T. Incidence and natural history of Mycobacterium avium complex infections in patients with advanced human immunodeficiency virus disease treated with zidovudine. Am Rev Respir Dis 1992;146:285--9. 235. Shanson DC, Dryden MS. Comparison of methods for isolating Mycobacterium avium intracellulare from blood of patients with AIDS. J Clin Pathol 1988;41:687--90. 236. Evans KD, Nakasome AS, Sutherland PA, de la Maza LM, Peterson EM. Identification of Mycobacterium tuberculosis and Mycobacterium--avium M. intracellulare directly from primary BACTEC cultures by using acridinium-ester--labelled DNA probes. J Clin Microbiol 1992;30: 2427--31. 237. Heifets L, Lindholm Levy P, Libonati J, et al. Radiometric broth macrodilution method for determination of minimal inhibitory concentrations (MIC) with Mycobacterium avium complex isolates: proposed guidelines. Denver, CO: National Jewish Center for Immunology and Respiratory Medicine, 1993. 238. Hafner R, Inderlied CB, Peterson DM, et al. Correlation of quantitative bone marrow and blood cultures in AIDS patients with disseminated Mycobacterium avium complex infection. J Infect Dis 1999;180:438--47. 239. Inderlied CB. Microbiology and minimum inhibitory concentration testing for Mycobacterium avium complex prophylaxis. Am J Med 1997;102:2--10. 240. Chaisson RE, Benson CA, Dube MP, et al. Clarithromycin therapy for bacteremic Mycobacterium avium complex disease. A randomized, double-blind, dose-ranging study in patients with AIDS. Ann Intern Med 1994;121:905--11. 241. Benson CA. Treatment of disseminated disease due to the Mycobacterium avium complex in patients with AIDS. Clin Infect Dis 1994;18 (Suppl 3):237--42. 242. Jacobson MA, Yajko D, Northfelt D, et al. Randomized, placebo-controlled trial of rifampin, ethambutol, and ciprofloxacin for AIDS patients with disseminated Mycobacterium avium complex infection. J Infect Dis 1993;168:112--9. 243. Kemper CA, Meng RC, Nussbaum J, et al. Treatment of Mycobacterium avium complex bacteremia in AIDS with a four drug oral regimen. Rifampin, ethambutol, clofazimine, and ciprofloxacin. Ann Intern Med 1992;116:466--72. 244. Shafran SD, Singer J, Zarowney DP, et al. A comparison of two regimens for the treatment of Mycobacterium avium complex bacteremia in AIDS: rifabutin, ethambutol, and clarithromycin versus rifampin, ethambutol, clofazimine, and ciprofloxacin. N Engl J Med 1996;335:377--83. 245. May T, Brel F, Beuscart C, et al. Comparison of combination therapy regimens for treatment of human immunodeficiency virus--infected patients with disseminated bacteremia due to Mycobacterium avium. Clin Infect Dis 1997;25:621--9. 246. Gordin FM, Sullam PM, Shafran SD, et al. A randomized, placebo-controlled study of rifabutin added to a regimen of clarithromycin and ethambutol for treatment of disseminated infection with Mycobacterium avium complex. Clin Infect Dis 1999;28:1080--5. 247. Dube MP, Sattler FR, Torriani FJ, et al. A randomized evaluation of ethambutol for prevention of relapse and drug resistance during treatment of Mycobacterium avium complex bacteremia with clarithromycin-based combination therapy. J Infect Dis 1997;176:1225--32. 248. Cohn DL, Fisher EJ, Peng GT, et al. A prospective randomized trial of four three-drug regimens in the treatment of disseminated Mycobacterium avium complex disease in AIDS patients: excess mortality associated with high-dose clarithromycin. Clin Infect Dis 1999;29:125--33. 249. Chaisson RE, Keiser P, Pierce M, et al. Clarithromycin and ethambutol with or without clofazimine for the treatment of bacteremic Mycobacterium avium complex disease in patients with HIV infection. AIDS 1997;11:311--7. 250. Masur H. Recommendations on prophylaxis and therapy for disseminated Mycobacterium avium complex disease in patients infected with the human immunodeficiency virus. N Engl J Med 1993;329:898--904. 251. Benson CA, Williams PL, Currier JS, et al. A prospective, randomized trial examining the efficacy and safety of clarithromycin in combination with ethambutol, rifabutin, or both for the treatment of disseminated Mycobacterium avium complex disease in persons with acquired immune deficiency syndrome. Clin Infect Dis 2003;37:1234--43. 252. Chiu J, Nussbaum J, Bozzette S, et al. Treatment of disseminated Mycobacterium avium complex infection in AIDS with amikacin, ethambutol, rifampin, and ciprofloxacin. Ann Intern Med 1990;113:358--61. 253. Aberg JA, Yajko DM, Jacobson MA. Eradication of AIDS-related disseminated Mycobacterium avium complex infection after 12 months of antimycobacterial therapy combined with highly active antiretroviral therapy. J Infect Dis 1998;178:1446--9. 254. Ward TT, Rimland D, Kauffman C, Huycke M, Evans TG, Heifets L. Randomized, open-label trial of azithromycin plus ethambutol vs. clarithromycin plus ethambutol as therapy for Mycobacterium avium complex bacteremia in patients with human immunodeficiency virus infection. Clin Infect Dis 1998;27:1278--85. 255. Dunne M, Fessel J, Kumar P, et al. A randomized, double-blind trial comparing azithromycin and clarithromycin in the treatment of disseminated Mycobacterium avium infection in patients with human immunodeficiency virus. Clin Infect Dis 2000;31: 1245--52. 256. Graves M, Salvato P, Thompson C. MAIC and the effect of prednisone on disease progression in AIDS patients [Abstract]. Presented at the 11th International Conference on AIDS, Vancouver, Canada, July 7--12, 1996. 257. Wormser GP, Horowitz H, Dworkin B. Low-dose dexamethasone as adjuvant therapy for disseminated Mycobacterium avium complex infections in AIDS patients. Antimicrob Agents Chemother 1994;38:2215--7. 258. Abbot Laboratories. Clarithromycin (biaxin) [package insert]. Abbott Park, IL: Abbott Laboratories, 1995. 259. Shafran SD, Deschenes J, Miller M, Phillips P, Toma E. Uveitis and pseudojaundice during a regimen of clarithromycin, rifabutin, and ethambutol. N Engl J Med 1994;330:438--9. 260. Hafner R, Bethel J, Power M, et al. Tolerance and pharmacokinetic interactions of rifabutin and clarithromycin in human immunodeficiency virus--infected volunteers. Antimicrob Agents Chemother 1998;42:631--9. 261. Heifets L, Mor N, Vanderkolk J. Mycobacterium avium strains resistant to clarithromycin and azithromycin. Antimicrob Agents Chemother 1993;37:2364--70. 262. Kemper CA, Bermudez L, Deresinski S. Immunomodulatory treatment of Mycobacterium avium complex bacteremia in patients with AIDS by use of recombinant granulocyte-macrophage colony--stimulating factor. J Infect Dis 1998;177:914--20. 263. Holland SM, Eisenstein DM, Kuhns DB, et al. Treatment of refractory disseminated nontuberculous mycobacterial infection with interferon gamma. N Engl J Med 1994;330:1348--55. 264. Nightingale SD, Cameron DW, Gordin FM, et al. Two controlled trials of rifabutin prophylaxis against Mycobacterium avium complex infection in AIDS. N Engl J Med 1993;329:828--33. 265. Benson CA, Williams PL, Cohn DL, et al. Clarithromycin or rifabutin alone or in combination for primary prophylaxis of Mycobacterium avium complex disease in patients with AIDS: a randomized, double-blind, placebo-controlled trial. J Infect Dis 2000;181:1289--97. 266. Havlir DV, Dube MP, Sattler FR, et al. Prophylaxis against disseminated Mycobacterium avium complex with weekly azithromycin, daily rifabutin, or both. N Engl J Med 1996; 335:392--8. 267. El-Sadr WM, Burman WJ, Grant LB, et al. Discontinuation of prophylaxis against Mycobacterium avium complex disease in HIV-infected patients who have a response to antiretroviral therapy. N Engl J Med 2000;342:1085--92. 268. Currier JS, Williams PL, Koletar SL, et al. Discontinuation of Mycobacterium avium complex prophylaxis in patients with antiretroviral therapy--induced increases in CD4+ cell count. A randomized, double-blind, placebo-controlled trial. Ann Intern Med 2000;133:493--503. 269. Stadnicki SW, Kessedjian M-J, Stadler J, Tachibana M. Preclinical reproductive and teratology studies with azithromycin. Oyo Yakuri/Pharmacometrics 1996;51:85--95. 270. United States pharmacopeial dispensing information. Clarithromycin. In: Drug information for the health care professional, Vol. I. Englewood, CO: Micromedex, 2001. 271. Einarson A, Phillips E, Mawji M, et al. A prospective controlled multicentre study of clarithromycin in pregnancy. Am J Perinatol 1998;15:523--5. 272. Wilton LV, Pearce GL, Martin RM, Mackay FJ, Mann RD. The outcomes of pregnancy in women exposed to newly marketed drugs in general practice in England. Br J Obstet Gynaecol 1998;105:882--9. 273. Caiaffa WT, Graham NM, Vlahov D. Bacterial pneumonia in adult populations with human immunodeficiency virus (HIV) infection. Am J Epidemiol 1993;138:909--22. 274. Wallace JM, Rao AV, Glassroth J, et al. Respiratory illness in persons with human immunodeficiency virus infection. Am Rev Respir Dis 1993;148 (6Pt 1):1523--9. 275. Redd SC, Rutherford GW 3rd, Sande MA, et al. The role of human immunodeficiency virus infection in pneumococcal bacteremia in San Francisco residents. J Infect Dis 1990;162:1012--7. 276. Gilks CF. Pneumococcal disease and HIV infection. Ann Intern Med 1993;118:393. 277. Boschini A, Smacchia C, Di Fine M, et al. Community-acquired pneumonia in a cohort of former injection drug users with and without human immunodeficiency virus infection: incidence, etiologies, and clinical aspects. Clin Infect Dis 1996;23:107--13. 278. Mundy LM, Auwaerter PG, Oldach D, et al. Community-acquired pneumonia: impact of immune status. Am J Respir Crit Care Med 1995;152(4 Pt 1):1309--15. 279. Hirschtick RE, Glassroth J, Jordan MC, et al. Bacterial pneumonia in persons infected with the human immunodeficiency virus. N Engl J Med 1995;333:845--51. 280. Park DR, Sherbin VL, Goodman MS, et al. The etiology of community-acquired pneumonia at an urban public hospital: influence of human immunodeficiency virus infection and initial severity of illness. J Infect Dis 2001;184:268--77. 281. Rimland D, Navin TR, Lennox JL, et al. Prospective study of etiologic agents of community-acquired pneumonia in patients with HIV infection. AIDS 2002;16:2361--2. 282. Schuchat A, Broome CV, Hightower A, Costa SJ, Parkin W. Use of surveillance for invasive pneumococcal disease to estimate the size of the immunosuppressed HIV-infected population. JAMA 1991;265:3275--9. 283. McEllistrem MC, Mendelsohn AB, Pass MA, et al. Recurrent invasive pneumococcal disease in individuals with human immunodeficiency virus infection. J Infect Dis 2002;185:1364--8. 284. Steinhart R, Reingold AL, Taylor F, Anderson G, Wenger JD. Invasive Haemophilus influenzae infections in men with HIV infection. JAMA 1992;268:3350--2. 285. Shepp DH, Tang IT, Ramundo MB, Kaplan MK. Serious Pseudomonas aeruginosa infection in AIDS. J Acquir Immune Defic Syndr 1994;7:823--31. 286. Selwyn PA, Pumerantz AS, Durante A, et al. Clinical predictors of Pneumocystis carinii pneumonia, bacterial pneumonia and tuberculosis in HIV-infected patients. AIDS 1998;12:885--93. 287. Bartlett JG, Dowell SF, Mandell LA, File TM Jr, Musher DM, Fine MJ. Practice guidelines for the management of community-acquired pneumonia in adults. Clin Infect Dis 2000;31:347--82. 288. Heffelfinger JD, Dowell SF, Jorgensen JH, et al. Management of community-acquired pneumonia in the era of pneumococcal resistance: a report from the Drug-Resistant Streptococcus pneumoniae Therapeutic Working Group. Arch Intern Med 2000;160:1399--408. 289. Celum CL, Chaisson RE, Rutherford GW, Barnhart JL, Echenberg DF. Incidence of salmonellosis in patients with AIDS. J Infect Dis 1987;156:998--1002. 290. Angulo FJ, Swerdlow DL. Bacterial enteric infections in persons infected with human immunodeficiency virus. Clin Infect Dis 1995;21(Suppl 1):84--93. 291. Nelson MR, Shanson DC, Hawkins DA, et al. Salmonella, Campylobacter and Shigella in HIV-seropositive patients. AIDS 1992;6:1495--8. 292. Casado JL, Valdezate S, Calderon C, et al. Zidovudine therapy protects against Salmonella bacteremia recurrence in human immunodeficiency virus--infected patients. J Infect Dis 1999;179:1553--6. 293. Snijders F, Kuijper EJ, de Wever B, van der Hoek L, Danner SA, Dankert J. Prevalence of Campylobacter-associated diarrhea among patients infected with human immunodeficiency virus. Clin Infect Dis 1997;24:1107--13. 294. Tee W, Mijch A. Campylobacter jejuni bacteremia in human immunodeficiency virus (HIV)-infected and non-HIV-infected patients: comparison of clinical features and review. Clin Infect Dis 1998;26:91--6. 295. Baer JT, Vugia DJ, Reingold AL, Aragon T, Angulo FJ, Bradford WZ. HIV infection as a risk factor for shigellosis. Emerg Infect Dis 1999;5:820--3. 296. Kristjansson M, Viner B, Maslow JN. Polymicrobial and recurrent bacteremia with Shigella in a patient with AIDS. Scand J Infect Dis 1994;26:411--6. 297. Tee W, Mijch A, Wright E, Yung A. Emergence of multidrug resistance in Campylobacter jejuni isolates from three patients infected with human immunodeficiency virus. Clin Infect Dis 1995;21:634--8. 298. Meier PA, Dooley DP, Jorgensen JH, Sanders CC, Huang WM, Patterson JE. Development of quinolone-resistant Campylobacter fetus bacteremia in human immunodeficiency virus--infected patients. J Infect Dis 1998;177:951--4. 299. Guerrant RL, Van Gilder T, Steiner TS, et al. Practice guidelines for the management of infectious diarrhea. Clin Infect Dis 2001;32:331--51. 300. Spach DH, Koehler JE. Bartonella-associated infections. Infect Dis Clin North Am 1998;12:137--155. 301. Koehler JE, Quinn FD, Berger TG, LeBoit PE, Tappero JW. Isolation of Rochalimaea species from cutaneous and osseous lesions of bacillary angiomatosis. N Eng J Med 1992;327:1625--31. 302. Houpikian P, Raoult D. Molecular phylogeny of the genus Bartonella: what is the current knowledge? FEMS Microbiol Lett 2001;200:1--7. 303. Koehler JE, Glaser CA, Tapero JW. Rochalimaea henselae infection. A new zoonosis with the domestic cat as reservoir. JAMA 1994;271:531--5. 304. Baron AL, Steinbach LS, LeBoit PE, Mills CM, Gee JH, Berger TG. Osteolytic lesions and bacillary angiomatosis in HIV infection: radiologic differentiation from AIDS-related Kaposi sarcoma. Radiology 1990;177:77--81. 305. Koehler JE, LeBoit PE, Egbert TG, Berger TG. Cutaneous vascular lesions and disseminated cat-scratch disease in patients with the acquired immunodeficiency syndrome (AIDS) and AIDS-related complex. Ann Intern Med 1988;109:449--55. 306. LeBoit PE, Berger TG, Egbert BM, Beckstead JH, Yen TS, Stoler MH. The histopathology and differential diagnosis of a pseudoneoplastic infection in patients with human immunodeficiency virus disease. Am J Surg Pathol 1989;13:909--20. 307. Regnery RL, Olson JG, Perkins BA, Bibb W. Serological response to Rochalimaea henselae antigen in suspected cat-scratch disease. Lancet 1992;339:1443--5. 308. Riley LE, Tuomala RE. Bacillary angiomatosis in a pregnant patient with acquired immunodeficiency syndrome. Obstet Gynecol 1992;79(5 Pt2):818--9. 309. Blocker ME, Levine WC, St. Louis ME. HIV prevalence in patients with syphilis, United States. Sex Transm Dis 2000;27:53--9. 310. Torian LV, Makki HA, Menzies IB, Murrill CS, Weisfuse IB. HIV infection in men who have sex with men, New York City Department of Health Sexually Transmitted Disease Clinics, 1990--1999: a decade of serosurveillance finds that racial disparities and associations between HIV and gonorrhea persist. Sex Trans Dis 2002;29:73--8. 312. Stolte I, Dukers NH, de Wit JB, Fennema JS, Coutinho RA. Increase in sexually transmitted infections among homosexual men in Amsterdam in relation to HAART. Sex Transm Infect 2001;77:184--6. 313. Brodine SK, Starkey MJ, Shaffer RA, et al. Diverse HIV-1 subtypes and clinical, laboratory and behavioral factors in a recently infected US military cohort. AIDS 2003;17:2521--7. 314. Golden MR, Marra CM, Holmes KK. Update on syphilis: resurgence of an old problem. JAMA. 2003;290:1510--4. 315. Calza L, Manfredi R, Marinacci G, Tadolini M, Fortunato L, Chiodo F. Efficacy of penicillin G benzathine as antimicrobial treatment of cutaneous secondary syphilis in patients with HIV infection [Letter]. J Chemother 2002;14:533--4. 316. Rompalo AM, Lawlor J, Seaman P, Quinn TC, Zenilman JM, Hook EW 3rd. Modification of syphilitic genital ulcer manifestations by coexistent HIV infection. Sex Transm Dis 2001;28:448--54. 317. Musher DM, Hamill RJ, Baughn RE. Effect of human immunodeficiency virus (HIV) infection on the course of syphilis and on the response to treatment. Ann Int Med 1990;113:872--81. 318. Radolf J, Kaplan R. Unusual manifestations of secondary syphilis and abnormal humoral immune response to Treponema pallidum antigens in a homosexual man with asymptomatic human immunodeficiency virus infection. J Am Acad Dermatol 1988;18(2 Pt 2):423--8. 319. CDC. Sexually transmitted diseases treatment guidelines 2002. MMWR 2002;51(No. RR-6):1--78. 320. Bayne LL, Schmidley JW, Goodin DS. Acute syphilitic meningitis. Its occurrence after clinical and serologic cure of secondary syphilis with penicillin G. Arch Neurol 1986;43:137--8. 321. Berry C, Hooton TM, Collier AC, Lukehart SA. Neurologic relapse after benzathine penicillin therapy for secondary syphilis in a patient with HIV infection. N Engl J Med 1987;316:1587--9. 322. Rompalo A, Joesoef MR, O'Donnell JA, et al. Clinical manifestations of early syphilis by HIV status and gender: results of the syphilis and HIV study. Sex Transm Dis 2001;28:158--65. 323. Wicher K, Horowitz HW, Wicher V. Laboratory methods of diagnosis of syphilis for the beginning of the third millennium. Microbes Infect 1999;1:1035--49. 324. Marra CM, Maxwell CL, Smith SL, et al. Cerebrospinal fluid abnormalities in patients with syphilis: association with clinical and laboratory features. J Infect Dis 2004;189:369--76. 325. Rolfs RT, Joesoef MR, Hendershot EF, et al. A randomized trial of enhanced therapy for early syphilis in patients with or without human immunodeficiency virus infection. N Engl J Med 1997;337: 307--14. 326. Gordon SM, Eaton ME, George R, et al. The response of symptomatic neurosyphilis to high-dose intravenous penicillin G in patients with human immunodeficiency virus infection. N Engl J Med 1994;331:1469--73. 327. Marra C, Longstreth WT, Maxwell CL, Lukehart SA. Resolution of serum and cerebrospinal fluid abnormalities after treatment of neurosyphilis. Influence of concomitant human immunodeficiency virus infection. Sex Transm Dis 1996;23:184--9. 328. Marra CM, Maxwell CL, Tantalo L, et al. Normalization of cerebrospinal fluid abnormalities after neurosyphylis therapy: does HIV status matter? Clin Infect Dis 2004;38:1001--6. 329. Genc M, Ledger W. Syphilis in pregnancy. Sex Transm Infect 2000;76:73--9. 330. Lee MJ, Hallmark RJ, Frenkel LM, Del Priore G. Maternal syphilis and vertical perinatal transmission of human immunodeficiency virus type-1 infection. Int J Gynecol Obstet 1998;63:247--52. 331. Wendel GD Jr, Sheffield JS, Hollier LM, Hill JB, Ramsey PS, Sanchez PJ. Treatment of syphilis in pregnancy and prevention of congenital syphilis. Clin Infect Dis 2002;35(Suppl 2):200--9. 332. Tess BH, Rodrigues LC, Newell ML, Dunn DT, Lago TD. Breastfeeding, genetic, obstetric and other risk factors associated with mother-to-child transmission of HIV-1 in São Paolo State, Brazil. AIDS 1998;12:513--20. 333. Bobat R, Coovadia H, Coutsoudis A, Moodley D. Determinants of mother-to-child transmission of human immunodeficiency virus type 1 infection in a cohort from Durban, South Africa. Pediatr Infect Dis J 1996;15:604--10. 334. Donders GG, Desmyter J, Hooft P, Dewet GH. Apparent failure of one injection of benzathine penicillin G for syphilis during pregnancy in human immunodeficiency virus--seronegative African women. Sex Transm Dis 1997;24:94--101. 335. Sheffield JS, Sanchez PJ, Morris G, et al. Congenital syphilis after maternal treatment for syphilis during pregnancy. Am J Obstet Gynecol 2002;186:569--73. 336. Klein VR, Cox SM, Mitchell MD, Wendel GD Jr. The Jarisch-Herxheimer reaction complicating syphilotherapy in pregnancy. Obstet Gynecol 1990;75(3 Pt 1):375--9. 337. Klein RS, Harris CA, Small CB, Moll B, Lesser M, Friedland GH. Oral candidiasis in high-risk patients as the initial manifestation of the acquired immunodeficiency syndrome. N Engl J Med 1984;311:354--8. 338. Rex JH, Rinaldi MG, Pfaller MA. Resistance of Candida species to fluconazole. Antimicrob Agents Chemother 1995;39:1--8. 339. Fichtenbaum CJ, Koletar S, Yiannoutsos C, et al. Refractory mucosal candidiasis in advanced human immunodeficiency virus infection. Clin Infect Dis 2000;30:749--56. 340. Maenza JR, Merz WG, Romagnoli MJ, Keruly JC, Moore RD, Gallant JE. Infection due to fluconazole-resistant Candida in patients with AIDS: prevalence and microbiology. Clin Infect Dis 1997;24:28--34. 341. Martins MD, Lozano-Chiu M, Rex JH. Point prevalence of oropharyngeal carriage of fluconazole-resistant Candida in human immunodeficiency virus--infected patients. Clin Infect Dis 1997,25:843--6. 342. Rex JH, Walsh TJ, Sobel JD, et al. Practice guidelines for the treatment of candidiasis. Infectious Diseases Society of America. Clin Infect Dis 2000;30:662--78. 343. Tiboni GM, Iammarrone E, Giampietro F, Lamonaca D, Bellati U, Di Ilio C. Teratological interaction between the bis-triazole antifungal agent fluconazole and the anticonvulsant drug phenytoin. Teratology 1999;59:81--7. 344. Tachibana M, Noguchi Y, Monro AM. Toxicology of fluconazole in experimental animals. In: Fromtling RA, ed. Recent trends in the discovery, development and evaluation of antifungal agents. Barcelona, Spain: JR Prous Science Publishers, S.A., 1987:93--102. 345. Pursley T, Blomqjist IK, Abraham J, Andersen HF, Bartley JA. Fluconazole-induced congenital anomalies in three infants. Clin Infect Dis 1996;22:336--40. 346. Aleck KA, Bartley DL. Multiple malformation syndrome following fluconazole use in pregnancy: report of an additional patient. Am J Med Genet 1997;72:253--6. 347. Inman W, Pearce G, Wilton L. Safety of fluconazole in the treatment of vaginal candidiasis. A prescription-event monitoring study, with special reference to the outcome of pregnancy. Eur J Clin Pharmacol 1994;46:115--8. 348. Mastroiacovo P, Mazzone T, Botto LD, et al. Prospective assessment of pregnancy outcomes after first-trimester exposure to fluconazole. Am J Obstet Gynecol 1996;175:1645--50. 349. Sorensen HT, Nielsen GL, Olesen C, et al. Risk of malformations and other outcomes in children exposed to fluconazole in utero. Br J Clin Pharmacol 1999;48:234--8. 350. Van Cauteren H, Lampo A, Vandenberghe J, et al. Safety aspects of oral antifungal agents. Br J Clin Pract 1990;71(Suppl):47--9. 351. Bar-Oz B, Moretti ME, Bishai R, et al. Pregnancy outcome after in utero exposure to itraconazole: a prospective cohort study. Am J Obstet Gynecol 2000;183:617--20. 352. Powderly WG. Cryptococcosis. In: AIDS Therapy. Dolin R, Masur H, Saag M, eds. New York, NY: Churchill Livingstone, 1999:400--11. 353. Mirza SA, Phelan M, Rimland D, et al. The changing epidemiology of cryptococcosis: an update from population-based active surveillance in 2 large metropolitan areas, 1992--2000. Clin Infect Dis 2003;36:789--94. 354. Powderly WG, Cloud GA, Dismukes WE, Saag MS. Measurement of cryptococcal antigen in serum and cerebrospinal fluid: value in the management of AIDS-associated cryptococcal meningitis. Clin Infect Dis 1994;18:789--92. 355. Van der Horst CM, Saag NS, Cloud GA, et al. Treatment of cryptococcal meningitis associated with the acquired immunodeficiency syndrome. N Engl J Med 1997;337:15--21. 356. Saag MS, Graybill JR, Larsen R et al. Practice guidelines for the management of cryptococcal meningitis. Clin Infect Dis 2000;30: 710--18. 357. Leenders AC, Reiss P, Portegies P, et al. Liposomal amphotericin B (AmBisome) compared with amphotericin B both followed by oral fluconazole in the treatment of AIDS-associated cryptococcal meningitis. AIDS 1997;11:1463--71. 358. Saag MS, Cloud GA, Graybill JR, et al. A comparison of itraconazole versus fluconazole as maintenance therapy for AIDS-associated cryptococcal meningitis. Clin Infect Dis 1999; 28:291--6. 359. Larsen RA, Bozzette SA, Jones BE, et al. Fluconazole combined with flucytosine for the treatment of cryptococcal meningitis in patients with AIDS. Clin Infect Dis 1994;19:741--747. 360. Graybill JR, Sobel J, Saag M, et al. Diagnosis and management of increased intracranial pressure in patients with AIDS and cryptococcal meningitis. Clin Infect Dis 2000;30:47--54. 361. Powderly WG, Saag MS, Cloud GA, et al. A controlled trial of fluconazole or amphotericin B to prevent relapse of cryptococcal meningitis in patients with the acquired immunodeficiency syndrome. N Engl J Med 1992;326:793--8. 362. Larsen RA. Editorial response: A comparison of itraconazole versus fluconazole as maintenance therapy for AIDS-associated cryptococcal meningitis. Clin Infect Dis 1999;28:297--8. 363. Chaube S, Murphy ML. The teratogenic effects of 5-fluorocytosine in the rat. Cancer Res 1969;9:554--7. 364. Wheat L. Histoplasmosis in the acquired immunodeficiency syndrome. Current Topics in Medical Mycology 1996;7:7--18. 365. Kaplan JE, Hanson D, Dworkin MS, et al. Epidemiology of human immunodeficiency virus--associated opportunistic infections in the United States in the era of highly active antiretroviral therapy. Clin Infect Dis 2000;30(Suppl 1):5--14. 366. Karimi K, Wheat LJ, Connolly P, et al. Differences in histoplasmosis in patients with acquired immunodeficiency syndrome in the United States and Brazil. J Infect Dis 2002;186:1655--60. 367. Wheat LJ, Connolly-Stringfield PA, Baker RL, et al. Disseminated histoplasmosis in the acquired immune deficiency syndrome: clinical findings, diagnosis and treatment, and review of the literature. Medicine 1990;69:361--74. 368. Williams B, Fojtasek M, Connolly-Stringfield P, Wheat J. Diagnosis of histoplasmosis by antigen detection during an outbreak in Indianapolis, Ind. Arch Pathol Lab Med 1994;118:1205--8. 369. Wheat JL. Current diagnosis of histoplasmosis. Trends Microbiol 2003;11:488--94. 370. Wheat J, Sarosi G, McKinsey D, et al. Practice guidelines for the management of patients with histoplasmosis. Clin Infect Dis 2000;30:688--95. 371. Johnson P, Wheat LJ, Cloud G, et al. Safety and efficacy of liposomal amphotericin B compared with conventional amphotericin B for induction therapy of histoplasmosis in patients with AIDS. Ann Intern Med 2002;137:105--9. 372. Wheat J, Hafner R, Korzun AH, et al. Itraconazole treatment of disseminated histoplasmosis in patients with the acquired immunodeficiency syndrome. Am J Med 1995;98:336--42. 373. Hecht FM, Wheat J, Korzun AH, et al. Itraconazole maintenance treatment for histoplasmosis in AIDS: a prospective, multicenter trial. J Acquir Immune Defic Syndr Hum Retrovirol 1997;16:100--7. 374. Galgiani JN, Ampel NM, Catanzaro A, Johnson RH, Stevens DA, Williams PL. Practice guideline for the treatment of coccidioidomycosis. Infectious Diseases Society of America. Clin Infect Dis 2000;30:658--61. 375. Galgiani JN, Catanzaro A, Cloud GA, et al. Comparison of oral fluconazole and itraconazole for progressive, nonmeningeal coccidioidomycosis. A randomized, double-blind trial. Ann Intern Med 2000;133:676--86. 376. Galgiani JN, Catanzaro A, Cloud GA, et al. Fluconazole therapy for coccidioidal meningitis. Ann Intern Med 1993;119:28--35. 377. Peterson CM, Schuppert K, Kelly PC, Pappagianis D. Coccidioidomycosis and pregnancy. Obstet Gynecol Surv 1993;48;149--56. 378. Powell BL, Drutz DJ, Huppert M, Sun SH. Relationship of progesterone- and estradiol-binding proteins in Coccidioides immitis to coccidioidal dissemination in pregnancy. Infect Immun 1983;40;478--85. 379. Holding KJ, Dworkin MS, Wan PC, et al. Aspergillosis among people infected with human immunodeficiency virus: incidence and survival. Adult and Adolescent Spectrum of HIV Disease Project. Clin Infect Dis 2000;31:1253--7. 380. Jabs DA, Van Natta ML, Kempen JH, et al. Characteristics of patients with cytomegalovirus retinitis in the era of highly active antiretroviral therapy. Am J Ophthalmol 2000;133:48--61. 381. Dieterich DT, Rahmin M. Cytomegalovirus colitis in AIDS: presentation in 44 patients and a review of the literature. J Acquir Immun Defic Syndr 1991;4(Suppl 1):29--35. 382. Arribas JR, Storch GA, Clifford DB, Tselis AC. Cytomegalovirus encephalitis. Ann Intern Med 1996;125:577--87. 383. Dodt KK, Jacobsen PH, Hofmann B, et al. Development of cytomegalovirus (CMV) disease may be predicted in HIV-infected patients by CMV polymerase chain reaction and the antigenemia test. AIDS 1997;11:F21--8. 384. Zurlo JJ, O'Neill D, Polis MA, et al. Lack of clinical utility of cytomegalovirus blood and urine cultures in patients with HIV infection. Ann Intern Med 1993;118:12--7. 385. Rodriguez-Barradas MC, Stool E, Musher DM, et al. Diagnosing and treating cytomegalovirus pneumonia in patients with AIDS. Clin Infect Dis 1996;23:76--81. 386. Arribas JR, Clifford DB, Fichtenbaum CJ, Commins DL, Powderly WG, Storch GA. Level of cytomegalovirus (CMV) DNA in cerebrospinal fluid of subjects with AIDS and CMV infection of the central nervous system. J Infect Dis 1995;172:527--31. 387. Wolf DG, Spector SA. Diagnosis of human cytomegalovirus central nervous system disease in AIDS patients by DNA amplification from cerebrospinal fluid. J Infect Dis 1992;166:1412--5. 388. Studies of Ocular Complications of AIDS Research Group in collaboration with the AIDS Clinical Trials Group. Foscarnet-ganciclovir cytomegalovirus retinitis trial. 4. Visual outcomes. Ophthalmology 1994;101:1250--61. 389. Musch DC, Martin DF, Gordon JF, Davis MD, Kuppermann BD. Treatment of cytomegalovirus retinitis with a sustained-release ganciclovir implant. N Engl J Med 1997;337:83--90. 390. Martin DF, Kuppermann BD, Wolitz RA, Palestine AG, Li H, Robinson CA. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. N Engl J Med 1999;340: 1063--70. 391. Martin DF, Sierra-Madero J, Walmsley S, et al. A controlled trial of valganciclovir as induction therapy for cytomegalovirus retinitis. N Engl J Med 2002;346;1119--26. 392. Kempen JH, Jabs DA, Wilson LA, Dunn JP, West SK, Tonascia JA. Risk of vision loss in patients with cytomegalovirus retinitis and the acquired immune deficiency syndrome. Arch Ophthalmol 2003;121:466--76. 393. Studies of Ocular Complications of AIDS Research Group. The AIDS Clinical Trials Group. The ganciclovir implant plus oral ganciclovir versus parenteral cidofovir for the treatment of cytomegalovirus retinitis in patients with acquired immunodeficiency syndrome. Am J Ophthalmol 2001;131:457--67. 394. Nguyen QD, Kempen JH, Bolton SG, Dunn JP, Jabs DA. Immune recovery uveitis in patients with AIDS and cytomegalovirus retinitis after highly active antiretroviral theapy. Am J Ophthalmol 2000;129:634--9. 395. Karavellas MP, Plummer DJ, Macdonald JC, et al. Incidence of immune recovery vitritis in cytomegalovirus retinitis patients following institution of successful highly active antiretroviral therapy. J Infec Dis 1999;179:697--700. 396. Robinson MR, Reed G, Csaky KG, Polis MA, Whitcup SM. Immune-recovery uveitis in patients with cytomegalovirus retinitis taking highly active antiretroviral therapy. Am J Ophthalmol 2000;130:49--56. 397. Karavellas MP, Song M, Macdonald JC, Freeman WR. Long-term posterior and anterior segment complications of immune recovery uveitis associated with cytomegalovirus retinitis. Am J Ophthalmol 2000;130:57--64. 398. Jabs DA, Wingard JR, de Bustros S, de Miranda P, Saral R, Santos GW. BW B759U for cytomegalovirus retinitis: intraocular drug penetration. Arch Ophthalmol 1986;104:1436--7. 399. Kupperman BD, Quiceno JI, Flores-Aguilar M, et al. Intravitreal ganciclovir concentration after intravenous administration in AIDS patients with cytomegalovirus retinitis: implications for therapy. J Infect Dis 1993;168:1506--9. 400. Arevalo JF, Gonzalez C, Capparelli EV, et al. Intravitreous and plasma concentrations of ganciclovir and foscarnet after intravenous therapy in patients with AIDS and cytomegalovirus retinitis. J Infect Dis 1995;172:951--6. 401. Marx JL, Kapusta MA, Patel SS, et al. Use of the ganciclovir implant in the treatment of recurrent cytomegalovirus retinitis. Arch Ophthalmol 1996;114:815--20. 402. Hatton MR, Duker JS, Reichel E, Morley MG, Puliafito CA. Treatment of relapsed cytomegalovirus retinitis with the sustained-release ganciclovir implant. Retina 1998;18:50--5. 403. The Studies of the Ocular Complications of AIDS Research Group in collaboration with the AIDS Clinical Trials Group. Combination foscarnet and ganciclovir therapy vs monotherapy for the treatment of relapsed cytomegalovirus retinitis in patients with AIDS. The cytomegalovirus retreatment trial. Arch Ophthalmol 1996;114: 23--33. 404. Jabs DA, Enger C, Dunn JP, Forman M. Cytomegalovirus retinitis and viral resistance: ganciclovir resistance. J Infect Dis 1998;177:770--3. 405. Jabs DA, Enger C, Forman M, Dunn JP. Incidence of foscarnet resistance and cidofovir resistance in patients treated for cytomegalovirus retinitis. Antimicrob Agents Chemother 1998;42:2240--4. 406. Jabs DA, Martin BK, Forman MS, et al., for the Cytomegalovirus Retinitis and Viral Resistance Study Group. Longitudinal observations on mutations conferring ganciclovir resistance in patients with acquired immunodeficiency syndrome and cytomegalovirus retinitis. Am J Ophthalmol 2001;132:700--10. 407. Chou S, Erice A, Jordan MC, et al. Analysis of the UL97 phosphotransferase coding sequence in clinical cytomegalovirus isolates and identification of mutations conferring ganciclovir resistance. J Infect Dis 1995;171:576--83. 408. Chou S, Guentzel S, Michels KR, Miner RC, Drew WL. Frequency of UL97 phosphotransferase mutations related to ganciclovir resistance in clinical cytomegalovirus isolates. J Infect Dis 1995;172: 239--42. 409. Smith IL, Cherrington JM, Jiles RE, Fuller MD, Freeman WR, Spector SA. High-level resistance of cytomegalovirus to ganciclovir is associated with alterations in both the UL97 and DNA polymerase genes. J Infect Dis 1997;176:69--77. 410. Jabs DA, Martin BK, Forman MS, et al. Mutations conferring ganciclovir resistance in a cohort of patients with acquired immunodeficiency syndrome and cytomegalovirus retinitis. J Infect Dis 2001;183;333--7. 411. Chou SW, Miner RC, Drew WL. A deletion mutation in region V of the cytomegalovirus DNA polymerase sequence confers multidrug resistance. J Infect Dis 2000;182;1765--8. 412. Wolf DG, Smith IL, Lee DJ, Freeman WR, Flores-Aguilar M, Spector SA. Mutations in human cytomegalovirus UL97 gene confer clinical resistance to ganciclovir and can be detected directly in patient plasma. J Clin Invest 1995;95:257--63. 413. The Vitravene Study Group. A randomized controlled clinical trial of intravitreous fomivirsen for treatment of newly diagnosed peripheral cytomegalovirus retinitis in patients with AIDS. Am J Ophthalmol 2002;133;467--74. 414. The Vitravene Study Group. Randomized dose-comparison studies of intravitreous fomivirsen for treatment of cytomegalovirus retinitis that has reactivated or is persistently active despite other therapies in patients with AIDS. Am J Ophthalmol 2002;133;475--83. 415. Jabs DA, Bolton SG, Dunn JP, Palestine AG. Discontinuing anti-cytomegalovirus therapy in patients with immune reconstitution after combination antiretroviral therapy. Am J Ophthalmol 1998;26: 817--22. 416. Vrabec TR, Baldassano VF, Whitcup SM. Discontinuation of maintenance therapy in patients with quiescent cytomegalovirus retinitis and elevated CD4+ counts. Ophthalmology 1998;105:1259--64. 417. Whitcup SM, Fortin E, Lindblad AS, et al. Discontinuation of anticytomegalovirus therapy in patients with HIV infection and cytomegalovirus retinitis. JAMA 1999;282:1633--7. 418. Tural C, Romeu J, Sirera G, et al. Long-lasting remission of cytomegalovirus retinitis without maintenance therapy in human immunodeficiency virus--infected patients. J Infect Dis 1998;177:1080--3. 419. Macdonald JC, Torriani FJ, Morse LS, Karavellas MP, Reed JB, Freeman WR. Lack of reactivation of cytomegalovirus (CMV) retinitis after stopping CMV maintenance therapy in AIDS patients with sustained elevations in CD4 T cells in response to highly active antiretroviral therapy. J Infect Dis 1998;177:1182--7. 420. Torriani FJ, Freeman WR, Macdonald JC, et al. CMV retinitis recurs after stopping treatment in virological and immunological failure of potent antiretroviral therapy. AIDS 2000;14:173--80. 421. Faqi AS, Klug A, Merker HJ, Chahoud I. Ganciclovir induces reproductive hazards in male rats after short-term exposure. Hum Exp Toxicol 1997;16:505--11. 422. Miller BW, Howard TK, Goss JA, Mostello DJ, Holcomb WL Jr, Brennan DC. Renal transplantation one week after conception. Transplantation 1995;60:1353--4. 423. Pescovitz MD. Absence of teratogenicity of oral ganciclovir used during early pregnancy in a liver transplant recipient. Transplantation 1999;67:758--9. 424. Alvarez-McLeod A, Havlik J, Drew KE. Foscarnet treatment of genital infection due to acyclovir-resistant herpes simplex virus type 2 in a pregnant patient with AIDS: case report. Clin Infect Dis 1999;29:937--8. 425. Gerber S, Hohlfeld P. Screening for infectious diseases. Childs Nerv Syst 2003;19:429--32. 426. Lipitz S, Achiron R, Zalel Y, Mendelson E, Tepperberg M, Gamzu R. Outcome of pregnancies with vertical transmission of primary cytomegalovirus infection. Obstet Gynecol 2002;100:428--33. 427. Stagno S, Pass RF, Cloud G, et al. Primary cytomegalovirus infection in pregnancy. Incidence, transmission to fetus, and clinical outcome. JAMA 1986;256:1904--8. 428. Yow MD, Williamson DW, Leeds LJ, et al. Epidemiologic characteristics of cytomegalovirus infection in mothers and their infants. Am J Obstet Gynecol 1998;158:1189--95. 429. Kovacs A, Schulchter M, Easley K, et al. Cytomegalovirus infection and HIV-1 disease progression in infants born to HIV-1--infected women. N Engl J Med 1999;341:77--84. 430. Quinn TC, Piot P, McCormick JB, et al. Serologic and immunologic studies in patients with AIDS in North America and Africa. The potential role of infectious agents as cofactors in human immunodeficiency virus infection. JAMA 1987;257:2617--21. 431. Mussi-Pinhata MM, Yamamoto AY, Figueiredo LT, Cervi MC, Duarte G. Congenital and perinatal cytomegalovirus infection in infants born to mothers infected with human immuno-deficiency virus. J Pediatr 1998;132:285--90. 432. Fleming DT, McQuillan GM, Johnson RE, et al. Herpes simplex virus type 2 in the United States, 1976 to1994. N Engl J Med 1997;337:1105--11. 433. Schacker T, Hu HL, Koelle DM, et al. Famciclovir for the suppression of symptomatic and asymptomatic herpes simplex virus reactivation in HIV-infected persons. Ann Intern Med 1998;128:21--8. 434. Corey L, Adams HG, Brown ZA, Holmes KK. Genital herpes simplex virus infections: clinical manifestations, course, and complications. Ann Intern Med 1983;98:958--72. 435. Meyers JD, Wade JC, Mitchell CD, et al. Multicenter collaborative trial of intravenous acyclovir for treatment of mucocutaneous herpes simplex virus infection in the immunocompromised host. Am J Med 1982;73:229--35. 436. Safrin S, Elbeik T, Phan L, Robinson D, Rush J, Elbaggari A, Mills J. Correlation between response to acyclovir and foscarnet therapy and in vitro susceptibility result for isolates of herpes simplex virus from human immunodeficiency virus--infected patients. Antimicrob Agents Chemother 1994;38:1246--50. 437. Balfour HH Jr. Antiviral drugs. N Engl J Med 1999;340:1255--68. 438. Feinberg JE, Hurwitz S, Cooper D, et. al. A randomized, double-blind trial of valacyclovir prophylaxis for cytomegalovirus disease in patients with advanced human immunodeficiency virus infection. J Infect Dis 1998;177;48--56. 439. Balfour HH Jr, Benson C, Braun J, et al. Management of acyclovir-resistant herpes simplex and varicella-zoster virus infections. J Acquir Immune Defic Syndr 1994;7:254--60. 440. Schacker T, Zeh J, Hu HL, Hill E, Corey L. Frequency of symptomatic and asymptomatic herpes simplex virus type 2 reactivations among human immunodeficiency virus--infected men. J Infect Dis 1998;178: 1616--22. 441. Reiff-Eldridge R, Heffner CR, Ephross SA, Tennis PS, White AD, Andrews EB. Monitoring pregnancy outcomes after prenatal drug exposure through prospective pregnancy registries: a pharmaceutical company commitment. Am J Obstet Gynecol 2000;182:159--63. 442. Kimberlin DF, Weller S, Whitley RJ, et al. Pharmacokinetics of oral valacyclovir and acyclovir in late pregnancy. Am J Obstet Gynecol 1998;179:846--51. 443. Hitti J, Watts DH, Burchett SK, et al. Herpes simplex virus seropositivity and reactivation at delivery among pregnant women infected with human immunodeficiency virus-1. Am J Obstet Gynecol 1997;177:450--4. 444. Augenbraun M, Feldman J, Chirgwin K, et al. Increased genital shedding of herpes simplex virus type 2 in HIV-seropositive women. Ann Intern Med 1995;123:845--7. 445. Engstrom RE, Jr., Holland GN, Margolis TP, et al. The progressive outer retinal necrosis syndrome. A variant of necrotizing herpetic retinopathy in patients with AIDS. Ophthalmology 1994;101: 1488--502. 446. Franco-Paredes C, Bellehemeur T, Merchant A, Sanghi P, DiazGranados C, Rimland D. Aseptic meningitis and optic neuritis preceding varicella-zoster progressive outer retinal necrosis in a patient with AIDS. AIDS 2002;16:1045--9. 447. Balfour HH, Jr., Bean B, Laskin OL, et al. Acyclovir halts progression of herpes zoster in immunocompromised patients. N Engl J Med 1983;308:1448--53. 448. Prober CG, Kirk LE, Keeney RE. Acyclovir therapy of chickenpox in immunosuppressed children---a collaborative study. J Pediatr 1982;101:622--5. 449. Carcao MD, Lau RC, Gupta A, Huerter H, Koren G, King SM. Sequential use of intravenous and oral acyclovir in the therapy of varicella in immunocompromised children. Pediatr Infect Dis J 1998;17:626--31. 450. Pastuszak AL, Ley M, Schick B, et al. Outcome after maternal varicella infection in the first 20 weeks of pregnancy. N Engl J Med 1994;330:901--5. 451. Gao SJ, Kingsley L, Hoover DR, et al. Seroconversion to antibodies against Kaposi's sarcoma--associated herpesvirus-related latent nuclear antigens before the development of Kaposi's sarcoma. N Engl J Med 1996;335:233--41. 452. Lennette ET, Blackbourn DJ, Levy JA. Antibodies to human herpesvirus type 8 in the general population and in Kaposi's sarcoma patients. Lancet 1996;348:858--61. 453. Cannon JS, Hamzeh F, Moore S, Nicholas J, Ambiner RF. Human herpesvirus 8-encoded thymidine kinase and phosphotransferase homologues confer sensitivity to ganciclovir. J Virol 1999;73: 4786--93. 454. Neyts J, De Clercq E. Antiviral drug susceptibility of human herpesvirus 8. Antimicrob Agents Chemother 1997;41:2754--6. 455. Kedes DH, Ganem D. Sensitivity of Kaposi's sarcoma--associated herpesvirus replication to antiviral drugs. Implications for potential therapy. J Clin Invest 1997;99:2082--6. 456. Ioannidis JP, Collier AC, Cooper DA, et al. Clinical efficacy of high-dose acyclovir in patients with human immunodeficiency virus infection: a meta-analysis of randomized individual patient data. J Infect Dis 1998;178:349--59. 457. Mocroft A, Youle M, Gazzard B, Morcinek J, Halai R, Phillips AN. Anti-herpesvirus treatment and risk of Kaposi's sarcoma in HIV infection. AIDS 1996;10:1101--5. 458. Glesby MJ, Hoover DR, Weng S, et al. Use of antiherpes drugs and the risk of Kaposi's sarcoma: data from the Multicenter AIDS Cohort Study. J Infect Dis 1996;173:1477--80. 459. Morfeldt L, Torssander J. Long-term remission of Kaposi's sarcoma following foscarnet treatment in HIV-infected patients. Scand J Infect Dis 1994;26:749--52. 460. Robles R, Lugo D, Gee L, Jacobson MA. Effect of antiviral drugs used to treat cytomegalovirus end-organ disease on subsequent course of previously diagnosed Kaposi's sarcoma in patients with AIDS. J Acquired Immune Defic Syndr Hum Retrovirol 1999;20:34--8. 461. Goedert JJ, Kedes DH, Ganem D. Antibodies to human herpesvirus 8 in women and infants born in Haiti and the USA [Letter]. Lancet 1999;349:1368. 462. Huang LM, Huang SY, Chen MY, et al. Geographical differences in human herpesvirus 8 seroepidemiology: a survey of 1,201 individuals in Asia. J Med Virol 2000;60:290--3. 463. Calabro ML, Gasperini P, Barbierato M, et al. A search for human herpesvirus 8 (HHV-8) in HIV-1 infected mothers and their infants does not suggest vertical transmission of HHV-8 [Letter]. Int J Cancer 2000;85:296--7. 464. Guitierrez-Ortega P, Heirro-Orozco S, Sanchez-Cisneros Cuevas F, Montano LF. Kaposi's sarcoma in a 6-day-old infant with human immunodeficiency virus. Arch Dermatol 1989;125:432--3. 465. McCarthy GA, Kampmann B, Novelli V, Miller RF, Mercey DE, Gibb D. Vertical transmission of Kaposi's sarcoma [Letter]. Arch Dis Child 1996;74:455--7. 466. Sitas F, Newton R, Boshoff C. Increasing probability of mother-to-child transmission of HHV-8 with increasing maternal antibody titer for HHV-8. N Engl J Med 1999;340:1923. 467. Mantina H, Kankasa C, Klaskala W, et al. Vertical transmission of Kaposi's sarcoma--associated herpesvirus. Int J Cancer 2001;94: 749--52. 468. Serraino D, Locatelli M, Songini M, et al. Human herpes virus-8 infection among pregnant women and their children: results from the Sardinia-IDDM Study 2 [Letter]. Int J Cancer 2001;91:740--1. 469. Gessain A, Mauclere P, Van Beveren M, et al. Human herpesvirus 8 primary infection occurs during childhood in Cameroon, Central Africa [Letter]. Int J Cancer 1999;81:189--92. 470. Bourboulia D, Whitby D, Boshoff C, et al. Serologic evidence for mother-to-child transmission of Kaposi sarcoma--associated herpesvirus infection. JAMA 1996;280:31--2. 471. Plancoulaine S, Abel L, van Beveren M, et al. Human herpesvirus 8 transmission from mother to child and between siblings in an endemic population. Lancet 2000;356:1062--5. 472. Marra CM, Rajicic N, Barker DE, et al. for the Adult AIDS Clinical Trials Group 363 Team. A pilot study of cidofovir for progressive multifocal leukoencephalopathy in AIDS. AIDS 2002;16:1791--7. 473. Lalezari JP, Holland GN, Kramer F, et al. Randomized, controlled study of the safety and efficacy of intravenous cidofovir for the treatment of relapsing cytomegalovirus retinitis in patients with AIDS. J Acquir Immune Defic Syndr Hum Retrovirol 1998;17:339--44. 474. Koutsky L. Epidemiology of genital human papillomavirus infection. Am J Med 1997;102:3--8. 475. Ho GY, Bierman R, Beardsley L, Chang CJ, Burk RD. Natural history of cervicovaginal papillomavirus infection in young women. N Engl J Med 1998;338:423--8. 476. Baken LA, Koutsky LA, Kuypers J, et al. Genital human papillomavirus infection among male and female sex partners: prevalence and type-specific concordance. J Infect Dis 1995;171:429--32. 477. Van Doornum GJ, Prins M, Juffermans LH, et al. Regional distribution and incidence of human papillomavirus infections among heterosexual men and women with multiple sexual partners: a prospective study. Genitourin Med 1994;70:240--6. 478. Chiasson MA, Ellerbrock TV, Bush TJ, Sun XW, Wright TC Jr. Increased prevalence of vulvovaginal condyloma and vulvar intraepithelial neoplasia in women infected with the human immunodeficiency virus. Obstet Gynecol 1997;89(5 Pt 1):690--4. 479. Conley LJ, Ellerbrock TV, Bush TJ, Chiasson MA, Sawo D, Wright TC. HIV-1 infection and risk of vulvovaginal and perianal condylomata acuminata and intraepithelial neoplasia: a prospective cohort study. Lancet 2002;359:108--13. 480. Gemignani M, Maiman M, Fruchter RG, Arrastia CD, Gibbon D, Ellison T. CD4 lymphocytes in women with invasive and preinvasive cervical neoplasia. Gynecol Oncol 1995;59:364--9. 481. Sun XW, Kuhn L, Ellerbrock TV, Chiasson MA, Bush TJ, Wright TC Jr. Human papillomavirus infection in women infected with the human immunodeficiency virus. N Engl J Med 1997;337:1343--9. 482. Palefsky JM, Minkoff H, Kalish LA, et al. Cervicovaginal human papillomavirus infection in human immunodeficiency virus-1 (HIV)--positive and high-risk HIV-negative women. J Natl Cancer Inst 1999;91:226--36. 483. Massad LS, Riester KA, Anastos KM, et al. Prevalence and predictors of squamous cell abnormalities in Papanicolaou smears from women infected with HIV-1. J Acquir Immune Defic Syndr Hum Retrovirol 1999;21:33--41. 484. Holly EA, Ralston ML, Darragh TM, Greenblatt RM, Jay N, Palefsky JM. Prevalence and risk factors for anal squamous intraepithelial lesions in women. J Natl Cancer Inst 2001;93:843--9. 485. Palefsky JM, Holly EA, Ralston ML, Jay N. Prevalence and risk factors for human papillomavirus infection of the anal canal in human immunodeficiency virus (HIV)--positive and HIV-negative men. J Infect Dis 1998;177:361--7. 486. Palefsky JM, Holly EA, Hogeboom CJ, et al. Virologic, immunologic, and clinical parameters in the incidence and progression of anal squamous intraepithelial lesions among HIV-positive and HIV-negative homosexual men. J Acquir Immune Defic Syndr Hum Retrovirol 1998;17:314--19. 487. Frisch M, Biggar RJ, Goedert JJ. Human papillomavirus--associated cancers in patients with human immunodeficiency virus infection and acquired immunodeficiency syndrome. J Natl Cancer Inst 2000;92:1500--10. 488. Wright TC, Jr, Cox JT, Massad LS, Twiggs LB, Wilkinson EJ. Consensus guidelines for the management of women with cervical cytological abnormalities. JAMA 2002;287:2120--9. 489. Beutner KR, Wiley DJ, Douglas JM, et al. Genital warts and their treatment. Clin Infect Dis 1999;28(Suppl 1):37--56. 490. Bonnez W, Elswick RK, Jr, Bailey-Farchione A, et al. Efficacy and safety of 0.5% podofilox solution in the treatment and suppression of anogenital warts. Am J Med 1994;96:420--5. 491. Tyring S, Edwards L, Cherry LK, et al. Safety and efficacy of 0.5% podofilox gel in the treatment of anogenital warts. Arch Dermatol 1998;134:33--8. 492. Beutner KR, Tyring SK, Trofatter KF, et al. Imiquimod, a patient-applied immune-response modifier for treatment of external genital warts. Antimicrob Agents Chemother 1998;42:789--94. 493. Edwards L, Ferenczy A, Eron L, et al. Self-administered topical 5% imiquimod cream for external anogenital warts. Human Papilloma Virus. Arch Dermatol 1998;134:25--30. 494. Gilson RJ, Shupack JL, Friedman-Kien AE, et al. A randomized, controlled, safety study using imiquimod for the topical treatment of anogenital warts in HIV-infected patients. AIDS 1999;13:2397--404. 495. Matteelli A, Beltrame A, Graifemberghi S, et al. Efficacy and tolerability of topical 1% cidofovir cream for the treatment of external anogenital warts in HIV-infected persons. Sex Transm Dis 2001;28:343--6. 496. Snoeck R, Bossens M, Parent D, et al. Phase II double-blind, placebo-controlled study of the safety and efficacy of cidofovir topical gel for the treatment of patients with human papillomavirus infection. Clin Infect Dis 2001;33:597--602. 497. Fruchter RG, Maiman M, Sedlis A, Bartley L, Camilien L, Arrastia CD. Multiple recurrences of cervical intraepithelial neoplasia in women with the human immunodeficiency virus. Obstet Gynecol 1996;87:338--44. 498 Chang GJ, Berry JM, Jay N, Palefsky JM, Welton ML. Surgical treatment of high-grade anal squamous intraepithelial lesions: a prospective study. Dis Colon Rectum 2002;45:453--8. 499. Wright TC, Koulos J, Schnoll F, et al. Cervical intraepithelial neoplasia in women infected with the human immunodeficiency virus: outcome after loop electrosurgical excision. Gynecol Oncology 1994;55:253--8. 500. Maiman M, Watts DH, Andersen, Clax P, Merino M, Kendall MA. Vaginal 5-fluorouracil for high-grade cervical dysplasia in HIV-infection: a randomized trial. Obstet Gynecol 1999;94:954--61. 501. Orr JW Jr., Barrett JM, Orr PF, Holloway RW, Holimon JL. The efficacy and safety of the cytobrush during pregnancy. Gynecol Oncol 1992;44:260--2. 502. Rivlin ME, Woodliff JM, Bowlin RB, et al. Comparison of cytobrush and cotton swab for Papanicolaou smears in pregnancy. J Reprod Med 1993;38:147--50. 503. Foster JC, Smith HL. Use of the cytobrush for Papanicalaou smear screens in pregnant women. J Nurse Midwifery 1996;41:211--7. 504. Creasman WT. Cancer and pregnancy. Ann N Y Acad Sci 2001;943:281--6. 505. Morrison EAB, Gammon MD, Goldberg GL, Vermund SH, Burk RD. Pregnancy and cervical infection with human papillomaviruses. Int J Gynecol Obstet 1996;54:125--30. 506. Kjelberg L, Hallmans G, Ahren A-M, et al. Smoking, diet, pregnancy and oral contraceptive use as risk factors for cervical intra-epithelial neoplasia in relation to human papillomavirus infection. Br J Cancer 2000;82:1332--8. 507. Shah K, Kashima H, Polk BF, Shah F, Abbey H, Abramson A. Rarity of cesarean delivery in cases of juvenile-onset respiratory papillomatosis. Obstet Gynecol 1986;68:795--9. 508. Fife KH, Katz BP, Brizendine EJ, Brown DR. Cervical human papillomavirus deoxyribonucleic acid persists throughout pregnancy and decreases in the postpartum period. Am J Obstet Gynecol 1999;180:1110--4. 509. Puranen MH, Yliskoski MH, Saarikoski SV, Syrjanen KJ, Syrjanen SM. Exposure of an infant to cervical human papillomavirus infection of the mother is common. Am J Obstet Gynecol 1997;176:1039--45. 510. Watts DH, Koutsky LA, Holmes KK, et al. Low risk of perinatal transmission of human papillomavirus: results from a prospective cohort study. Am J Obstet Gynecol 1998;178:365--73. 511. Tseng CJ, Liang CC, Soong YK, Pao CC. Perinatal transmission of human papillomavirus in infants: relationship between infection rate and mode of delivery. Obstet Gynecol 1998;91:92--6. 512. Tenti P, Zappatore R, Migliora P, Spinello A, Belloni C, Carnevali L. Perinatal transmission of human papillomavirus from gravidas with latent infections. Obstet Gynecol 1999;93:475--9. 513. Garfein RS, Vlahov D, Galai N, Doherty MC, Nelson KE. Viral infections in short-term injection drug users: the prevalence of the hepatitis C, hepatitis B, human immunodeficiency, and human T- lymphotropic viruses. Am J Public Health 1996;86:655--61. 514. Van Ameijden EJ, Van den Hoek JA, Mientjes GH, Coutinho RA. A longitudinal study on the incidence and transmission patterns of HIV, HBV and HCV infection among drug users in Amsterdam. Eur J Epidemiol 1993;9:255--62. 515. Makris M, Preston FE, Triger DR, et al. Hepatitis C antibody and chronic liver disease in haemophilia. Lancet 1990;335:1117--9. 516. Rumi MG, Colombo M, Gringeri A, Mannucci PM. High prevalence of antibody to hepatitis C virus in multitransfused hemophiliacs with normal transaminase levels. Ann Intern Med 1990;112: 379--80. 517. Alter MJ, Hadler SC, Judson FN, et al. Risk factors for acute non-A, non-B hepatitis in the United States and association with hepatitis C virus infection. JAMA 1990;264:2231--5. 518. Conry-Cantilena C, VanRaden M, Gibble J, et al. Routes of infection, viremia, and liver disease in blood donors found to have hepatitis C virus infection. N Engl J Med 1996;334:1691--6. 519. Gordon SC, Patel AH, Kulesza GW, Barnes RE, Silverman AL. Lack of evidence for the heterosexual transmission of hepatitis C. Am J Gastroenterol 1992;87:1849--51. 520. Brettler DB, Mannucci PM, Gringeri A, et al. The low risk of hepatitis C virus transmission among sexual partners of hepatitis C-infected hemophilic males: an international, multicenter study. Blood 1992;80:540--3. 521. Zanetti AR, Tanzi E, Paccagnini S, et al. Mother-to-infant transmission of hepatitis C virus. Lancet 1995;345:289--91. 522. Thomas DL, Villano SA, Riester KA, et al. Perinatal transmission of hepatitis C virus from human immunodeficiency virus type 1--infected mothers. J Infect Dis 1998;177:1480--8. 523. Sherman KE, Rouster SD, Chung RT, Rajicic N. Hepatitis C Virus prevalence among patients infected with human immunodeficiency virus: a cross-sectional analysis of the US adult AIDS Clinical Trials Group. Clin Infect Dis 2002;34:831--7. 524. Alter HJ, Seeff LB. Recovery, persistence, and sequelae in hepatitis C virus infection: a perspective on long-term outcome. Semin Liver Dis 2000;20:17--35. 525. Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. Lancet 1997;349: 825--32. 526. Kenny-Walsh E. Clinical outcomes after hepatitis C infection from contaminated anti-D immune globulin. N Engl J Med 1999;340: 1228--33. 527. Vogt M, Lang T, Frosner G, et al. Prevalence and clinical outcome of hepatitis C infection in children who underwent cardiac surgery before the implementation of blood-donor screening. N Engl J Med 1999;341:866--70. 528. Seeff LB, Hollinger FB, Alter HJ, et al. Long-term mortality and morbidity of transfusion-associated non-A, non- B, and type C hepatitis: a National Heart, Lung, and Blood Institute collaborative study. Hepatology 2001;33:455--63. 529. Darby SC, Ewart DW, Giangrande PL, et al. Mortality from liver cancer and liver disease in haemophilic men and boys in UK given blood products contaminated with hepatitis C. Lancet 1997;350: 1425--31. 530. Lesens O, Deschenes M, Steben M, Belanger G, Tsoukas CM. Hepatitis C virus is related to progressive liver disease in human immunodeficiency virus--positive hemophiliacs and should be treated as an opportunistic infection. J Infect Dis 1999;179:1254--8. 531. Ragni MV, Belle SH. Impact of human immunodeficiency virus infection on progression to end-stage liver disease in individuals with hemophilia and hepatitis C virus infection. J Infect Dis 2001;183: 1112--5. 532. Benhamou Y, Bochet M, Di Martino V, et al. Liver fibrosis progression in human immunodeficiency virus and hepatitis C virus coinfected patients. Hepatology 1999;30:1054--8. 533. Romeo R, Rumi MG, Donato MF, et al. Hepatitis C is more severe in drug users with human immunodeficiency virus infection. J Viral Hepat 2000;7:297--301. 534. Pol S, Fontaine H, Carnot F, et al. Predictive factors for development of cirrhosis in parenterally acquired chronic hepatitis C: a comparison between immunocompetent and immunocompromised patients. J Hepatol 1998;29:12--9. 535. Graham CS, Baden LR, Yu E, et al. Influence of human immunodeficiency virus infection on the course of hepatitis C virus infection: a meta-analysis. Clin Infect Dis 2001;33:562--9. 536. Wright TL, Hollander H, Pu X, et al. Hepatitis C in HIV-infected patients with and without AIDS: prevalence and relationship to patient survival. Hepatology 1994;20:1152--5. 537. Dorrucci M, Pezzotti P, Phillips AN, Lepri AC, Rezza G. Coinfection of hepatitis C virus with human immunodeficiency virus and progression to AIDS. J Infect Dis 1995;172:1503--8. 538. Sabin CA, Telfer P, Phillips AN, Bhagani S, Lee CA. The association between hepatitis C virus genotype and human immunodeficiency virus disease progression in a cohort of hemophilic men. J Infect Dis 1997;175:164--8. 539. Piroth L, Grappin M, Cuzin L, et al. Hepatitis C virus co-infection is a negative prognostic factor for clinical evolution in human immunodeficiency virus--positive patients. J Viral Hepat 2000;7:302--8. 540. Piroth L, Bourgeois C, Dantin S, et al. Hepatitis C virus (HCV) genotype does not appear to be a significant prognostic factor in HIV-HCV--coinfected patients [Letter]. AIDS 1999;13:523--4. 541. Rosenberg PM, Farrell JJ, Abraczinskas DR, Graeme-Cook FM, Dienstag JL, Chung RT. Rapidly progressive fibrosing cholestatic hepatitis---hepatitis C virus in HIV coinfection. Am J Gastroenterol 2002;97:478--83. 542. Gholson CF, Morgan K, Catinis G, et al. Chronic hepatitis C with normal aminotransferase levels: a clinical histologic study. Am J Gastroenterol 1997;92:1788--92. 543. Inglesby TV, Rai R, Astemborski J, et al. A prospective, community-based evaluation of liver enzymes in individuals with hepatitis C after drug use. Hepatology 1999;29:590--6. 544. Thio CL, Nolt KR, Astemborski J, Vlahov D, Nelson KE, Thomas DL. Screening for hepatitis C virus in human immunodeficiency virus--infected individuals. J Clin Microbiol 2000;38:575--7. 545. Bejarano PA, Koehler A, Sherman KE. Second opinion pathology in liver biopsy interpretation. Am J Gastroenterol 2001;96:3158--64. 546. Imbert-Bismut F, Ratziu V, Pieroni L, Charlotte F, Benhamou Y, Poynard T . Biochemical markers of liver fibrosis in patients with hepatitis C virus infection: a prospective study. Lancet 2001;357: 1069--75. 547. McAfee JH, Keeffe EB, Lee RG, Rosch J. Transjugular liver biopsy. Hepatology 1992;15:726--32. 548. Fried MW. Management of hepatitis C in the hemophilia patient. Am J Med 1999;107:85S--89S. 549. Pessione F, Degos F, Marcellin P, et al. Effect of alcohol consumption on serum hepatitis C virus RNA and histological lesions in chronic hepatitis C. Hepatology 1998;27:1717--22. 550. Gow PJ, Pillay D, Mutimer D. Solid organ transplantation in patients with HIV infection. Transplantation 2001;72:177--81. 551. DiMartino V, Thevenot T, Boyer N, Degos F, MArcellin P. Serum alanine transaminase level is a good predictor of response to interferon alfa therapy for chronic hepatitis B in human immunodeficiency virus--infected patients [Letter]. Hepatology 2000;31:1030--1. 552. Nishiguchi S, Kuroki T, Nakatani S, et al. Randomised trial of effects of interferon-alpha on incidence of hepatocellular carcinoma in chronic active hepatitis C with cirrhosis. Lancet 1995;346:1051--5. 553. Shiffman ML, Hofmann CM, Contos MJ, et al. A randomized, controlled trial of maintenance interferon therapy for patients with chronic hepatitis C virus and persistent viremia. Gastroenterology 1999;117:1164--72. 554. Manns MP, McHutchison JG, Gordon SC, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 2001;358:958--65. 555. Zylberberg H, Pol S. Characteristics and treatment of hepatitis C virus infection in HIV- coinfected subjects. AIDS Patient Care STDS 1998;12:11--9. 556. Nasti G, Di Gennaro G, Tavio M, et al. Chronic hepatitis C in HIV infection: feasibility and sustained efficacy of therapy with interferon alfa-2b and ribavirin. AIDS 2001;15:1783--7. 557. Landau A, Batisse D, Piketty C, et al. Long-term efficacy of combination therapy with interferon-alpha 2b and ribavirin for severe chronic hepatitis C in HIV-infected patients. Aids 2001;15:2149--55. 558. Sauleda S, Juarez A, Esteban JI, et al. Interferon and ribavirin combination therapy for chronic hepatitis C in human immunodeficiency virus--infected patients with congenital coagulation disorders. Hepatology 2001;34:1035--40. 559. Poynard T, Marcellin P, Lee SS, et al. Randomised trial of interferon alpha2b plus ribavirin for 48 weeks or for 24 weeks versus interferon alpha2b plus placebo for 48 weeks for treatment of chronic infection with hepatitis C virus. Lancet 1998;352:1426--32. 560. McHutchison JG, Gordon SC, Schiff ER, et al. Interferon alfa-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. N Engl J Med 1998;339:1485--92. 561. Soriano V, Garcia-Samaniego J, Bravo R, et al. Interferon alpha for the treatment of chronic hepatitis C in patients infected with human immunodeficiency virus. Clin Infect Dis 1996;23:585--91. 562. Vogt MW, Hartshorn KL, Furman PA, et al. Ribavirin antagonizes the effect of azidothymidine on HIV replication. Science 1987;235: 1376--9. 563. Hoggard PG, Kewn S, Barry MG, Khoo SH, Back DJ. Effects of drugs on 2',3'-dideoxy-2',3'-didehydrothymidine phosphorylation in vitro. Antimicrob Agents Chemother 1997;41:1231--6. 564. Baba M, Pauwels R, Balzarini J, Herdewijn P, De Clercq E, Desmyter J. Ribavirin antagonizes inhibitory effects of pyrimidine 2',3'- dideoxynucleosides but enhances inhibitory effects of purine 2',3'- dideoxynucleosides on replication of human immunodeficiency virus in vitro. Antimicrob Agents Chemother 1987;31:1613--7. 565. Gervais A, Bacq Y, Bernauau J, et al. Decrease in serum ALT and increase in serum HCV RNA during pregnancy in women with chronic hepatitis C. J Hepatol 2000;32:293--99. 566. Conte D, Fraquelli M, Prati D, Colucci A, Minola E. Prevalence and clinical course of chronic hepatitis C virus (HCV) infection and rate of HCV vertical transmission in a cohort of 15,250 pregnant women. Hepatology 2000;31:751--5. 567. Paternoster DM, Santarossa C, Grella P, et al. Viral load in HCV RNA-positive pregnant women. Am J Gastroenterol 2001;96:2751--4. 568. Hiratsuka M, Minkami H, Koshizuka S, Sato I. Administration of interferon-alpha during pregnancy: effects on fetus. J Perinat Med 2000;28:372--6. 569. Trotter JF, Zygmunt AJ. Conception and pregnancy during interferon-alpha therapy for chronic hepatitis C. J Clin Gastroenterol 2001;32:76--8. 570. Mubarak AAS, Kakil IR, Awidi A, et al. Normal outcome of pregnancy in chronic myeloid leukemia treated with interferon-alpha in 1st trimester: report of 3 cases and review of the literature. Am J Hematol 2002;69:115--8. 571. American College of Obstetricians and Gynecologists. Viral hepatitis in pregnancy. Educational bulletin no. 248, July 1998. 572. Johnson EM. The effects of ribavirin on development and reproduction: a critical review of published and unpublished studies in experimental animals. J Am Coll Toxicol 1990;9:551--61. 573. Yeung LT, King SM, Roberts EA. Mother-to-infant transmission of hepatitis C virus. Hepatology 2001;34:223--9. 574. Tajiri H, Miyoshi Y, Shunpei F, et al. Prospective study of mother-to-infant transmission of hepatitis C virus. Pediatr Infect Dis J 2001;20:10--4. 575. Gibb DM, Goodall RL, Dunn DT, et al. Mother-to-child transmission of hepatitis C virus: evidence for preventable peripartum transmission. Lancet 2000;356:904--7. 576. European Paediatric Hepatitis C Virus Network. Effects of mode of delivery and infant feeding on the risk of mother-to-child transmission of hepatitis C virus. Brit J Obstet Gynecol 2001;108:371--7. 577. Hershow RC, Riester KA, Lew J, et al. Increased vertical transmission of human immunodeficiency virus from hepatitis C virus--coinfected mothers. J Infect Dis 1997;176:414--20. 578. Lee WM. Hepatitis B virus infection. N Engl J Med 1997;337: 1733--45. 579. Levine OS, Vlahov D, Koehler J, Cohn S, Spronk AM, Nelson KE. Seroepidemiology of hepatitis B virus in a population of injecting drug users. Association with drug injection patterns. Am J Epidemiol 1995;142:331--41. 580. Beasley RP, Trepo C, Stevens CE, Szmuness W. The e antigen and vertical transmission of hepatitis B surface antigen. Am J Epidemiol 1977;105:94--8. 581. Rodriguez-Mendez ML, Gonzalez-Quintela A, Aguilera A, Barrio E. Prevalence, patterns, and course of past hepatitis B virus infection in intravenous drug users with HIV-1 infection. Am J Gastroenterol 2000;95:1316--22. 582. Scharschmidt BF, Held MJ, Hollander HH, et al. Hepatitis B in patients with HIV infection: relationship to AIDS and patient survival. Ann Intern Med 1992;117:837--8. 583. Homann C, Krogsgaard K, Pedersen C, Andersson P, Nielsen JO. High incidence of hepatitis B infection and evolution of chronic hepatitis B infection in patients with advanced HIV infection. J Acquir Immune Defic Syndr 1991;4:416--20. 584. Bodsworth NJ, Cooper DA, Donovan B. The influence of human immunodeficiency virus type 1 infection on the development of the hepatitis B carrier state. J Infect Dis 1991;163:1138--40. 585. Hadler SC, Judson FN, O'Malley PM, et al. Outcome of hepatitis B virus infection in homosexual men and its relation to prior human immunodeficiency virus infection. J Infect Dis 1991;163:454--9. 586. Houssett C, Pol S, Carnot F, et al. Interactions between human immunodeficiency virus-1, hepatitis delta virus and hepatitis B virus infections in 260 chronic carriers of hepatitis B virus. Hepatology 1992;15:578--83. 587. Colin JF, Cazals-Hatem D, Loriot MA, et al. Influence of human immunodeficiency virus infection on chronic hepatitis B in homosexual men. Hepatology 1999;29:1306--10. 588. Gilson RJ, Hawkins AE, Beecham MR, et al. Interactions between HIV and hepatitis B virus in homosexual men: effects on the natural history of infection. AIDS 1997;11:597--606. 589. Thio CL, Seaberg EC, Skolasky R Jr, et al. HIV-1, hepatitis B virus, and risk of liver-related mortality in the Multicenter Cohort Study (MACS). Lancet 2002;360:1921--6. 590. Grob P, Jilg W, Bornhak H, et al. Serological pattern "anti-HBc alone": report on a workshop. J Med Virol 2000;62:450--5. 591. Hofer M, Joller-Jemelka HI, Grob PJ, Luthy R, Opravil M. Frequent chronic hepatitis B virus infection in HIV-infected patients positive for antibody to hepatitis B core antigen only. Eur J Clin Microbiol Infect Dis 1998;17:6--13. 592. Silva AE, McMahon BJ, Parkinson AJ, Sjogren MH, Hoofnagle JH, Di Bisceglie AM. Hepatitis B virus DNA in persons with isolated antibody to hepatitis B core antigen who subsequently received hepatitis B vaccine. Clin Infect Dis 1998;26:895--7. 593. Lok AS, Lai CL, Wu PC. Prevalence of isolated antibody to hepatitis B core antigen in an area endemic for hepatitis B virus infection: implications in hepatitis B vaccination programs. Hepatology 1988;8:766--70. 594. Altfeld M, Rockstroh JK, Addo M, et al. Reactivation of hepatitis B in a long-term anti-HBs-positive patient with AIDS following lamivudine withdrawal. J Hepatol 1998;29:306--9. 595. Bessesen M, Ives D, Condreay L, Lawrence S, Sherman KE. Chronic active hepatitis B exacerbations in human immunodeficiency virus--infected patients following development of resistance to or withdrawal of lamivudine. Clin Infect Dis 1999;28:1032--5. 596 Perrillo RP. Acute flares in chronic hepatitis B: the natural and unnatural history of an immunologically mediated liver disease. Gastroenterology 2001;120:1009--22. 597. Sulkowski MS, Thomas DL, Chaisson RE, Moore RD. Elevated liver enzymes following initiation of antiretroviral therapy. JAMA 2000;283: 2526--7. 598. Lok AS, McMahon BJ. Chronic hepatitis B. Hepatology 2001;34: 1225--41. 599. Di Bisceglie AM, Rustgi VK, Hoofnagle JH, Dusheiko GM, Lotze MT. NIH conference. Hepatocellular carcinoma. Ann Intern Med 1988;108:390--401. 600. Neilsen GA, Bodsworth NJ, Watts N. Response to hepatitis A vaccination in human immunodeficiency virus--infected and --uninfected homosexual men. J Infect Dis. 1997;176:1064--7. 601. Wong DK, Yim C, Naylor CD, et al. Interferon alfa treatment of chronic hepatitis B: randomized trial in a predominantly homosexual male population. Gastroenterology 1995;108:165--71. 602. Krogsgaard K. The long-term effect of treatment with interferon-alpha 2a in chronic hepatitis B. J Viral Hepat 1998;5:389--97. 603. Cooksley WG, Piratvisuth T, Lee SD, et al. Peginterferon alpha-2a (40 kDa): an advance in the treatment of hepatitis B e antigen--positive chronic hepatitis B. J Viral Hepat 2003;10:298--305. 604. Lok AS, Heathcote EJ, Hoofnagle JH. Management of hepatitis B: 2000---summary of a workshop. Gastroenterology 2001;120: 1828--53. 605. Janssen HL, Gerken G, Carreno V, et al. Interferon alfa for chronic hepatitis B infection: increased efficacy of prolonged treatment. Hepatology 1999;30:238--43. 606. Marcellin P, Boyer N, Colin JF, et al. Recombinant alpha interferon for chronic hepatitis B in anti-HIV positive patients receiving zidovudine. Gut 1993;34(Suppl 2):106. 607. Visco G, Alba L, Grisetti S, et al. Zidovudine plus interferon alfa-2b treatment in patients with HIV and chronic active viral hepatitis. Gut 1993;34(Suppl 2):107--8. 608. Zylberberg H, Jiang J, Pialoux G, et al. Alpha-interferon for chronic active hepatitis B in human immunodeficiency virus--infected patients. Gastroenterol Clin Biol 1996;20:968--71. 609. Dore GJ, Cooper DA, Barrett C, Goh LE, Thakrar B, Atkins M. Dual efficacy of lamivudine treatment in human immunodeficiency virus/hepatitis B virus--coinfected persons in a randomized, controlled study (CAESAR). J Infect Dis 1999;180:607--13. 610. Tassopoulos NC, Volpes R, Pastore G, et al. Efficacy of lamivudine in patients with hepatitis B e antigen--negative/hepatitis B virus DNA--positive (precore mutant) chronic hepatitis B. Hepatology 1999;29: 889--96. 611. Barbaro G, Zechini F, Pellicelli AM, et al. Long-term efficacy of interferon alpha-2b and lamivudine in combination compared to lamivudine monotherapy in patients with chronic hepatitis B. An Italian multicenter, randomized trial. J Hepatol 2001;35:406--11. 612. Schalm SW, Heathcote J, Cianciara J, et al. Lamivudine and alpha interferon combination treatment of patients with chronic hepatitis B infection: a randomised trial. Gut 2000;46:562--8. 613. Matthews GV, Pillay D, Cane P, Ratcliffe D, Gazzard B, Nelson M. Failure of combination therapy with lamivudine and famciclovir following lamivudine monotherapy for hepatitis B virus infection in patients coinfected with human immunodeficiency virus-1. Clin Infect Dis 2001;33:2049--54. 614. Rayes N, Seehofer D, Hopf U, et al. Comparison of famciclovir and lamivudine in the long-term treatment of hepatitis B infection after liver transplantation. Transplantation. 2001;71:96--101. 615. de Man RA, Marcellin P, Habal F, et al. A randomized, placebo-controlled study to evaluate the efficacy of 12-month famciclovir treatment in patients with chronic hepatitis B e antigen--positive hepatitis B. Hepatology 2000;32:413--7. 616. Honkoop P, De Man RA, Niesters HG, Zondervan PE, Schalm SW. Acute exacerbation of chronic hepatitis B virus infection after withdrawal of lamivudine therapy. Hepatology 2000;32:635--9. 617. Benhamou Y, Bochet M, Thibault V, et al. Long-term incidence of hepatitis B virus resistance to lamivudine in human immunodeficiency virus--infected patients. Hepatology 1999;30:1302--6. 618. Lai CL, Chien RN, Leung NWY, et al. A one-year trial of lamivudine for chronic hepatitis B. N Engl J Med 1998;339:61--8. 619. Dienstag JL, Schiff ER, Wright TL, et al. Lamivudine as initial treatment for chronic hepatitis B in the United States. N Engl J Med 1999;341:1256--63. 620. Leung NW, Lai CL, Chang TT, et al. Extended lamivudine treatment in patients with chronic hepatitis B enhances hepatitis B e antigen seroconversion rates: results after 3 years of therapy. Hepatology 2001;33:1527--32. 621. Antiretroviral Pregnancy Registry Steering Committee. Antiretroviral Pregnancy Registry international interim report for 1 January 1989 through 31 July 2003. Wilmington, NC: Registry Coordinating Center, 2003. 622. Benhamou Y, Bochet M, Thibault V, et al. Safety and efficacy of adefovir dipivoxil in patients co-infected with HIV-1 and lamivudine-resistant hepatitis B virus: an open-label pilot study [Letter]. Lancet 2001;358:718--23. 623. van Nunen AB, de Man RA, Heijtink RA, Niesters HG, Schalm SW. Lamivudine in the last 4 weeks of pregnancy to prevent perinatal transmission in highly viremic chronic hepatitis B patients. J Hepatol 2000;32:1040--1. 624. Supparatpinyo K, Khamwan C, Baosoung V, Nelson KE, Sirisanthana T. Disseminated Penicillium marneffei infection in Southeast Asia. Lancet 1994;344:110--3. 625. Clezy K, Sirisanthana T, Sirisanthana V, Brew B, Cooper DA. Late manifestations of HIV in Asia and the Pacific. AIDS 1994;8 (Suppl 2):35--43. 626. Kantipong P, Panich V, Pongsurachet V, Watt G. Hepatic penicilliosis in patients without skin lesions. Clin Infect Dis 1998;26:1215--7. 627. Sirisanthana T, Supparatpinyo K, Perriens J, Nelson KE. Amphotericin B and itraconazole for treatment of disseminated Penicillium marneffei infection in human immunodeficiency virus-infected patients. Clin Infect Dis 1998;26:1107--10. 628. Supparatipinyo K, Perriens J, Nelson KE, Sirisanthana T. A controlled trial of itraconazole to prevent relapse of Penicillium marneffei infection in patients infected with the human immunodeficiency virus. N Engl J Med 1998;339:1739--43. 629. Pearson RD, Sousa AQ. Clinical spectrum of leishmaniasis. Clin Infect Dis 1996;22:1--13. 630. Alvar J, Canavale C, Guitierrez-Solar B, et al. Leishmania and human immunodeficiency virus coinfection: the first 10 years. Clin Microbiol Rev 1997;10:298--319. 631. Tortajada C, Perez-Cuevas B, Moreno A, et al. Highly active antiretroviral therapy (HAART) modifies the incidence and outcome of visceral leishmanaisis in HIV-infected patients [Letter]. J Acquir Immune Defic Syndr. 2002;30:364--6. 632. Rosenthal E, Tempesta S, del Giudice P, et al. Declining incidence of visceral leishmaniasis in HIV-infected individuals in the era of highly active antiretroviral therapy. AIDS 2001;15:1184--5. 633. Pintado V, Martin-Rabadan P, Riveraml ML, Moreno S, Bouza E. Visceral leishmaniasis in human immunodeficiency virus (HIV)--infected and non-HIV-infected patients. A comparative study. Medicine (Baltimore) 2001;80:54--73. 634. Rosenthal E, Marty P, del Giudice P, et al. HIV and Leishmania coinfection: a review of 91 cases with focus on atypical locations of Leishmania. Clin Infect Dis 2000;31:1093--5. 635. Mota-Sasaki MG, Matsumo MC, Schmitz Ferreira ML, Machado MP. Cutaneous leishmaniasis coinfection in AIDS patients: case report and literature review. Braz J Infect Dis 1997;1:142--4. 636. Pizzuto M, Piazza M, Senese D, et al. Role of PCR in diagnosis and prognosis of visceral leishmaniasis in patients co-infected with HIV-1. J Clin Microbiol 2001;39:357--61. 637. Salotra P, Sreenivas G, Pogue GP, et al. Development of a species-specific PCR assay for detection of Leishmania donovani in clinical samples from patients with kala-azar and post-kala-azar dermal leishmaniasis. J Clin Microbiol 2001;39:849--54. 638. Gramiccia M, Gradoni L, Troiani M. HIV-Leishmania co-infections in Italy. Isoenzyme characterization of Leishmania causing visceral leishmaniasis in HIV patients. Trans R Soc Trop Med Hyg 1992;86: 161--3. 639. Laguna F, Lopez-Velez R, Pulido F, et al. Treatment of visceral leishmanaisis in HIV infected patients: a randomized trial comparing meglumine antimoniate with amphotericin B. AIDS 1999;13: 1063--9. 640. Laguna F, Videla S, Jimenez-Mejias ME, et al. Amphotericin B lipid complex versus meglumine antimoniate in the treatment of visceral leishmaniasis in patients infected with HIV: a randomized pilot study. J Antimicrob Chemother 2003;52:464--8. 641. Cahn P, Badaro R, Freilij H. Other parasitic infections. In: Crowe S, Hoy J, Mills J, eds. Management of the HIV-infected patient. London, UK: Martin Dunitz, Ltd., 2001. 642. Badaro R, Nascimento C, Carvalho JS, et al. Granulocyte-macrophage colony-stimulating factor in combination with pentavalent antimony for the treatment of visceral Leishmaniasis. Eur J Clin Microbiol Infect Dis 1994;13(Suppl 2):23--8. 643. Mishra M, Biswas UK, Jhadn, Khan AB. Amphotericin versus pentamidine in antimony-unresponsive kala-azar. Lancet 1992;340:1256--7. 644. Torre-Cisneros J, Villanueva JL, Kindelan JM, Jurado R, Sanchez-Guijo P. Successful treatment of antimony-resistant visceral leishmaniasis with liposomal amphotericin B in patients infected with human immunodeficiency virus. Clin Infect Dis 1993;17:625--7. 645. Russo R, Nigro LC, Minniti S, et al. Visceral leishmaniasis in HIV infected patients: treatment with high dose liposomal amphotericin B (AmBisome). J Infect 1996;32:133--7. 646. Canora-Lebrato J, Troncoso-Garcia E, Escobar T, Hernandez-Quero J. Treatment of visceral leishmaniasis in HIV patients with a new regimen of liposomal amphotericin [letter]. Med Clin (Barc) 2001;116:395. 647. Badaro R, Johnson WD Jr. The role of interferon-gamma in the treatment of visceral and diffuse cutaneous leishmaniasis. J Infect Dis 1993;167(Suppl 1):13--7. 648. Sundar S, Jha TK, Thakur CP, et al. Oral miltefosine for Indian visceral leishmaniasis. N Engl J Med. 2002;347:1739--46 649. Ribera E, Ocana I, de Otero J, Cortes E, Gasser I, Pahissa A. Prophylaxis of visceral leishmaniasis in human immunodeficiency virus--infected patients. Am J Med 1996;100:496--501. 650. López-Vélez R, Videla S, Márquez M, et al. Amphotericin B lipid complex versus no-treatment in the secondary prophylaxis of visceral leishmaniasis in HIV-infected patients. J Antimicrob Chemother 2004;53:540--3. 651. Perez-Molina JA, Lopez-Velez R, Montilla P, Guerrero A. Pentamidine isethionate as secondary prophylaxis against visceral leishmaniasis in HIV-positive patients [Letter]. AIDS. 1996;10:237--8. 652. Berenguer J, Cosin J, Miralles P, Lopez JC, Padilla B. Discontinuation of secondary anti-leishmania prophylaxis in HIV-infected patients who have responded to highly active antiretroviral therapy. AIDS 2000;14:2946--8. 653. Casado JL, Lopez-Veles R, Pintado V, Quereda C, Antela A, Moreno S. Relapsing visceral leishmaniasis in HIV-infected patients undergoing successful protease inhibitor therapy. Eur J Clin Microbiol Infect Dis 2001;20:202--5. 654. Ridgway LP, Karnofsky DA. The effects of metals on the chick embryo: toxicity and production of abnormalities in development. Ann N Y Acad Sci 1952;55:203--15. 655. Rossi F, Acampora C, Vacca C, Maione S, Matera MG, Servodio R, Marmo E. Prenatal and postnatal antimony exposure in rats: effect on vasomotor reactivity development of pups. Teratogenesis Carcinog Mutagen 1987;7:491--6. 656. James LF, Lazar VA, Binns W. Effects of sublethal doses of certain minerals on pregnant ewes and fetal development. Am J Vet Res 1966;27:132--5. 657. Utili R, Rambaldi A, Tripodi MF, Andreanna A. Visceral leishmaniasis during pregnancy treated with meglumine antimoniate. Infection 1995;23:182--3. 658. Gradoni L, Gaeta GB, Pellizzer G, Maisto A, Scalone A. Mediterranean visceral leishmaniasis in pregnancy. Scand J Infect Dis 1994;26:627--9. 659. Meinecke CK, Schottelius J, Oskam L, Fleischer B. Congenital transmission of visceral leishmaniasis (kala azar) from an asymptomatic mother to her child. Pediatrics 1999;104:65--9. 660. Benard G, Duarte AJ. Paracoccidioidomycosis: a model for evaluation of the effects of human immunodeficiency virus infection on the natural history of endemic tropical diseases. Clin Infect Dis 2000;31:1032--9. 661. Goldani LZ, Sugar AM. Paracoccidioidomycosis and AIDS: an overview. Clin Infect Dis 1995;21:1275--81. 662. Nishioka S de A. Paracoccidioidomycosis and AIDS [Letter]. Clin Infect Dis 1996;22:1132--3. 663. Lortholary O, Denning DW, Dupont B. Endemic mycoses: a treatment update. J Antimicrob Chemother 1999;43:321--31. 664. Sorvillo FJ, Leib LE, Seidel P, Kerndt P, Turner J, Ash LR. Epidemiology of isosporiasis among persons with acquired immunodeficiency syndrome in Los Angeles County. Am J Trop Med Hyg 1995;53: 656--9. 665. Pape JW, Verdier RI, Johnson WD Jr. Treatment and prophylaxis of Isospora belli infections in patients with the acquired immunodeficiency syndrome. New Engl J Med 1989;320:1044--7. 666. Weiss LM, Perlman DC, Sherman J, Tanowitz H, Wittner M. Isospora belli infection: treatment with pyrimethamine. Ann Int Med 1988;109:474--5. 667. Verdier FJ, Fitzgerald DW, Johnson DW, Papp JW. Trimethoprim-sulfamethoxazole compared with ciprofloxacin for treatment and prophylaxis of Isospora belli and Cyclospora cayetanensis infection in HIV-infected patients. A randomized, controlled trial. Ann Int Med 2000;132:885--8. 668. Gaska JA, Tietze KJ, Cosgrove EM. Unsuccessful treatment of enteritis due to Isospora belli with spiramycin: a case report. J Infect Dis 1985;152:1336--8. 669. Musey KL, Chidiac C, Beaucaire G, Houriez S, Fourrier A. Effectiveness of roxithromycin for treating Isospora belli infection [Letter]. J Infect Dis 1988;158:646. 670. Limson-Pobre RN, Merrick S, Gruen D, Soave R. Use of diclazuril for the treatment of isosporiasis in patients with AIDS [Letter]. Clin Infect Dis 1995;20:201--2. 671. Romero-Cabello R, Guerrero KR, Munoz-Garcia MR, Geyne Cruz A. Nitazoxanide for the treatment of intestinal protozoan and helminthic infections in Mexico. Trans R Soc Trop Med Hyg 1997;91:701--3. 672. Dionisio D, Sterrantino G, Meli M, Leoncini F, Orsi A, Nicoleeti P. Treatment of isosporiasis with combined albendazole and ornidazole in patients with AIDS [Letter]. AIDS 1996;10:1301--2. 673. Raynaud F, Horvath C. Folate deficiency and congenital malformations induced by pyrimethamine in the rat. Reprod Nutr Dev 1994;34:461--71. 674. Matsui D. Prevention, diagnosis, and treatment of fetal toxoplasmosis. Clin Perinatol 1994;21:675--89. 675. Kirchhoff LV. American trypanosomiasis (Chagas' Disease)---a tropical disease now in the United States. N Engl J Med 1993:9:639--44. 676. Villalba R, Fornes G, Alvarez M, et al. Acute Chagas' disease in a recipient of a bone marrow transplant in Spain: case report. Clin Infect Dis 1992;14:594--5. 677. Grant IH, Gold JW, Wittener M, et al. Transfusion-associated acute Chagas disease acquired in the United States. Ann Intern Med 1989;111:849--51 678. Cahn P, Belloso W, Murillo J, Prada Trujillo G. AIDS in Latin America. Infect Dis Clin North Am 2000;14:185--209. 679. Kohl S, Pickering LK, Frankel LS, Yaeger RG. Reactivation of Chagas' disease during therapy of acute lymphocytic leukemia. Cancer 1982;50:827--8. 680. Leiguarda R, Roncoroni A, Taratuto AL, et al. Acute CNS infection by Trypanosoma cruzi in immunosuppressed patients. Neurology 1990;40:850--1. 681. Silva N, O'Bryan L, Medeiros E, et al. Trypanosoma cruzi meningoencephalitis in HIV-infected patients. J Acquir Immune Defic Syndr Hum Retrovirol 1999;20:342--9. 682. Rocha A, de Meneses AC, da Silva A, et al. Pathology of patients with Chagas' disease and acquired immunodeficiency syndrome. Am J Trop Med Hyg 1994;50:261--8. 683. Freilij H, Altcheh J, Muchinik G. Perinatal human immunodeficiency virus infection and congenital Chagas' disease. Pediatr Infect Dis J 1995;14:161--2. 684. Freilij H, Altcheh J. Congenital Chagas' disease: diagnostic and clinical aspects. Clin Infect Dis 1995;21:551--5. 685. Strout RG. A method for concentrating hemoflagellates J Parasitol 1962;48:100. 686. Schenone H, Alfaro E, Rojas A. Basis and yield of the xenodiagnosis in human Chagas infection. Bol Chil Parasitol 1974;29:24--6. 687. Ferreira AW. Serological diagnosis. In: Wendel S, ed. Chagas disease: its impact on transfusion and clinical medicine. ISBT: São Paulo, Brazil, 1992. 688. Di Pentima MC, Hwang LY, Skeeter CM, Edwards MS. Prevalence of antibody to Trypanosoma cruzi in pregnant Hispanic women in Houston. Clin Infect Dis 1999;28:1281--5. 689. Hernandez-Matheson IM, Frankowski RF, Held B. Foeto-maternal morbidity in the presence of antibodies to Trypanosoma cruzi. Trans R Soc Trop Med Hyg 1983;77:405--11. 690. Bittencourt AL. Possible risk factors for vertical transmission of Chagas' disease. Rev Inst Med Trop São Paulo 1992;34:403--8. 691. Gorla NB, Ledesma OS, Barbieri GP, Larripa IB. Assessment of cytogenetic damage in chagasic children treated with benznidazole. Mutat Res 1988;206:217--20. 692. Gorla NB, Ledesma OS, Barbieri GP, Larripa IB. Thirteenfold increase of chromosomal aberrations non-randomly distributed in chagasic children treated with nifurtimox. Mutat Res 1989;224: 263--7. 693. deToranzo EG, Masana M, Castro JA. Administration of benznidazole, a chemotherapeutic agent against Chagas' disease, to pregnant rats. Covalent binding of reactive metabolites to fetal and maternal proteins. Arch Int Pharmacodyn Ther 1984;272:17--23.
Chairs: Henry Masur, M.D., National Institutes of Health, Bethesda, Maryland; Jonathan E. Kaplan, M.D., CDC, Atlanta, Georgia; King K. Holmes, M.D., Ph.D., University of Washington, Seattle, Washington. Project Director/Co-Chair: Constance A. Benson, M.D., University of Colorado Health Sciences Center, Denver, Colorado. Contributors: Judith Aberg, M.D., Washington University, St. Louis, Missouri; O.C. Abraham, M.D., CMC Hospital, Vellore Tamil Nada, India; Neil Ampel, M.D., Southern Arizona VA Health Care System, Tuscon, Arizona; Jean Anderson, M.D., Johns Hopkins University, Baltimore, Maryland; Roberto Badaro, M.D., Universidade Federal da Bahia, Salvador, Bahia, Brazil; A. Cornelius Baker, Whitman Walker Clinic, Washington, DC. Henry Balfour, M.D., University of Minnesota, Minneapolis, Minnesota; David Barr, Lindesmith Center, New York, New York; John G. Bartlett, M.D., Johns Hopkins University, Baltimore, Maryland; Mary T. Bassett, M.D., The Rockefeller Foundation, Southern Africa Regional Office, Harare, Zimbabwe; John E. Bennett, M.D., National Institutes of Health, Bethesda, Maryland; William Bower, M.D., CDC, Atlanta, Georgia; Douglas Brust, M.D., Columbia University, New York, New York; William Burman, M.D., Denver Public Health Department, Denver, Colorado; Pedro Cahn, M.D., Fundación Huesped, Buenos Aires, Argentina; Victoria Cargill, M.D., National Institutes of Health, Bethesda, Maryland; Kenneth Castro, M.D., CDC, Atlanta, Georgia; Judith Currier, M.D., UCLA Care Center, Los Angeles, California; Lawrence Deyton, M.D., U.S. Department of Veterans Affairs, Washington, DC; William Duncan, M.D., National Institutes of Health, Rockville, Maryland; Robert Eisinger, Ph.D., National Institutes of Health, Bethesda, Maryland; Judith Falloon, M.D., National Institutes of Health, Bethesda, Maryland; Judith Feinberg, M.D., University of Cincinnati, Cincinnati, Ohio; Kenneth Fife, M.D., Ph.D., Indiana University, Indianapolis, Indiana; Timothy Flanigan, M.D., Miriam Hospital, Providence, Rhode Island; Hansjakob Furrer, M.D., University Hospital, Bern, Switzerland; Mark Goldberger, M.D., U.S. Food and Drug Administration, Rockville, Maryland; Fred Gordin, M.D., Veterans Administration Medical Center, Washington, DC; Paul Griffiths, M.D., Royal Free and University College Medical School, London, United Kingdom; Richard Hafner, M.D., National Institutes of Health, Rockville, Maryland; Diane Havlir, M.D., University of California, San Diego, California; Andrzeg Horban, M.D., Ph.D., Centrum Diagnostyki I Terapii, Warsaw, Poland; Douglas Jabs, M.D., Johns Hopkins University School of Medicine, Baltimore, Maryland; Pacharee Kantipong, M.D., Chiang Rai Regional Hospital, Chaing Rai, Thailand; Elly T Katabira, M.D., Makerere University, Kampala, Uganda; Ram Koppaka, M.D., Ph.D., CDC, Atlanta, Georgia; Joseph Kovacs, M.D., National Institutes of Health, Bethesda, Maryland; Catherine Leport, M.D., Groupe Hospitalier Bichat---Claude Bernard, Paris, France; Joseph Lovato, National Association of People with AIDS, Washington, DC; Jens Lundgren, M.D., Hvidore Hospital, Hvidore, Denmark; Michael Marco, Treatment Action Group, New York, New York; Christina Marra, M.D., University of Washington, Seattle, Washington; Jose Miro, M.D., University of Barcelona, Barcelona, Spain; Lynne Mofenson, M.D., National Institutes of Health, Rockville, Maryland; Theodore Nash, M.D., National Institutes of Health, Bethesda, Maryland; James Oleske, M.D., University of Medicine and Dentistry of New Jersey, Newark, New Jersey; Joseph O'Neill, M.D., U.S. Department of State, Washington, D.C.; Joel Palefsky, M.D., University of California, San Francisco, California; Alice Pau, Pharm.D., National Institutes of Health, Bethesda, Maryland; Marion Peters, M.D., University of California, San Francisco, California; Michael Polis, M.D., National Institutes of Health, Bethesda, Maryland; William Powderly, M.D., Washington University, St. Louis, Missouri; Peter Reiss, M.D., University of Amsterdam, Amsterdam, Netherlands; Renee Ridzon, M.D., CDC, Atlanta, Georgia; Paul E. Sax, M.D., Brigham and Women's Hospital, Boston, Massachusetts; Leonard Seeff, M.D., National Institutes of Health, Bethesda, Maryland; Kent Sepkowitz, M.D., Memorial Sloan Kettering Cancer Center, New York, New York; Leslie Serchuck, M.D., National Institutes of Health, Rockville, Maryland; Kenneth Sherman, M.D., Ph.D., University of Cincinnati, College of Medicine, Cincinnati, Ohio; Jack Sobel, M.D., Wayne State University/Detroit Medical Center, Detroit, Michigan; Kathleen Squires, M.D., University of Southern California, Los Angeles, California; Mark Sulkowski, M.D., Johns Hopkins University, Baltimore, Maryland; Michael Tapper, M.D., Lenox Hill Hospital, New York, New York; Amalio Telenti, M.D., Ph.D., Centre Hospitalier Universitaire Vaudois, Lausanne, Switzerland; Chloe Thio, M.D., Johns Hopkins University, Baltimore, Maryland; David Thomas, M.D., Johns Hopkins University, Baltimore, Maryland; Ruth E. Tuomala, M.D., Brigham and Women's Hospital, Boston, Massachusetts; Russell Van Dyke, M.D., Tulane Health Sciences Center, New Orleans, Louisiana; D. Heather Watts, M.D., National Institutes of Health, Rockville, Maryland; Louis Weiss, M.D., MPH, Albert Einstein College of Medicine, Bronx, New York; L. Joseph Wheat, M.D., Mira Vista Labs, Indianapolis, Indiana. Box  Return to top. Table 1
All MMWR HTML versions of articles are electronic conversions from ASCII text into HTML. This conversion may have resulted in character translation or format errors in the HTML version. Users should not rely on this HTML document, but are referred to the electronic PDF version and/or the original MMWR paper copy for the official text, figures, and tables. An original paper copy of this issue can be obtained from the Superintendent of Documents, U.S. Government Printing Office (GPO), Washington, DC 20402-9371; telephone: (202) 512-1800. Contact GPO for current prices. **Questions or messages regarding errors in formatting should be addressed to mmwrq@cdc.gov.Page converted: 12/13/2004 |
|||||||||
This page last reviewed 12/13/2004
|