Volume 30, Number 3—March 2024
Dispatch
Highly Pathogenic Avian Influenza A(H5N1) Virus Clade 2.3.4.4b in Domestic Ducks, Indonesia, 2022
Hendra Wibawa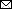 , Putut Eko Wibowo, Arif Supriyadi, Lestari Lestari, Jessiaman Silaban, Aziz Ahmad Fuadi, Anna Januar Fiqri, Retno Wulan Handayani, Sri Handayani Irianingsih, Zaza Fahmia, Herdiyanto Mulyawan, Syafrison Idris, and Nuryani Zainuddin
, Putut Eko Wibowo, Arif Supriyadi, Lestari Lestari, Jessiaman Silaban, Aziz Ahmad Fuadi, Anna Januar Fiqri, Retno Wulan Handayani, Sri Handayani Irianingsih, Zaza Fahmia, Herdiyanto Mulyawan, Syafrison Idris, and Nuryani Zainuddin
Figure

Figure. Phylogenetic analysis of the hemagglutinin gene of highly pathogenic avian influenza A(H5N1) clade 2.3.4.4b viruses isolated from domestic ducks during outbreaks in South Kalimantan, Indonesia, in April 2022 and July 2023 compared with reference sequences. Bold font indicates the viruses isolated from duck farms in this study. Letters at right indicate subclades. Evolutionary history was inferred by using the maximum-likelihood method and best-fit general time reversible plus gamma distribution 4 substitution model involving 67 hemagglutinin H5 sequences from the GISAID database (http://www.gisaid.org); a total of 1,656 positions were in the final dataset. Scale bar indicates nucleotide substitutions per site.
Page created: January 08, 2024
Page updated: February 22, 2024
Page reviewed: February 22, 2024
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.