Erythema Migrans Caused by Borrelia spielmanii, France
Pascal del Giudice
, Fabienne Freychet, Lora Kopec, Florence Fenollar, Carole Eldin, Marine Velin, Thomas Hubiche, Didier Raoult, and Oleg Mediannikov
Author affiliations: Dermatology Infectiology Unit, CH Fréjus-Saint-Raphaël, France (P. del Giudice, M. Velin, T. Hubiche); Dermatology private practice, Nice, France (F. Freychet); MEPHI, IHU Méditerranée infection, IRD, Aix Marseille Université, Marseille, France (L. Kopec, F. Fenollar, C. Eldin, D. Raoult, O. Mediannikov); VITROME, IHU Méditerranée infection, IRD, Aix Marseille Université, Marseille (L. Kopec, F. Fenollar, C. Eldin, D. Raoult, O. Mediannikov)
Main Article
Figure 2
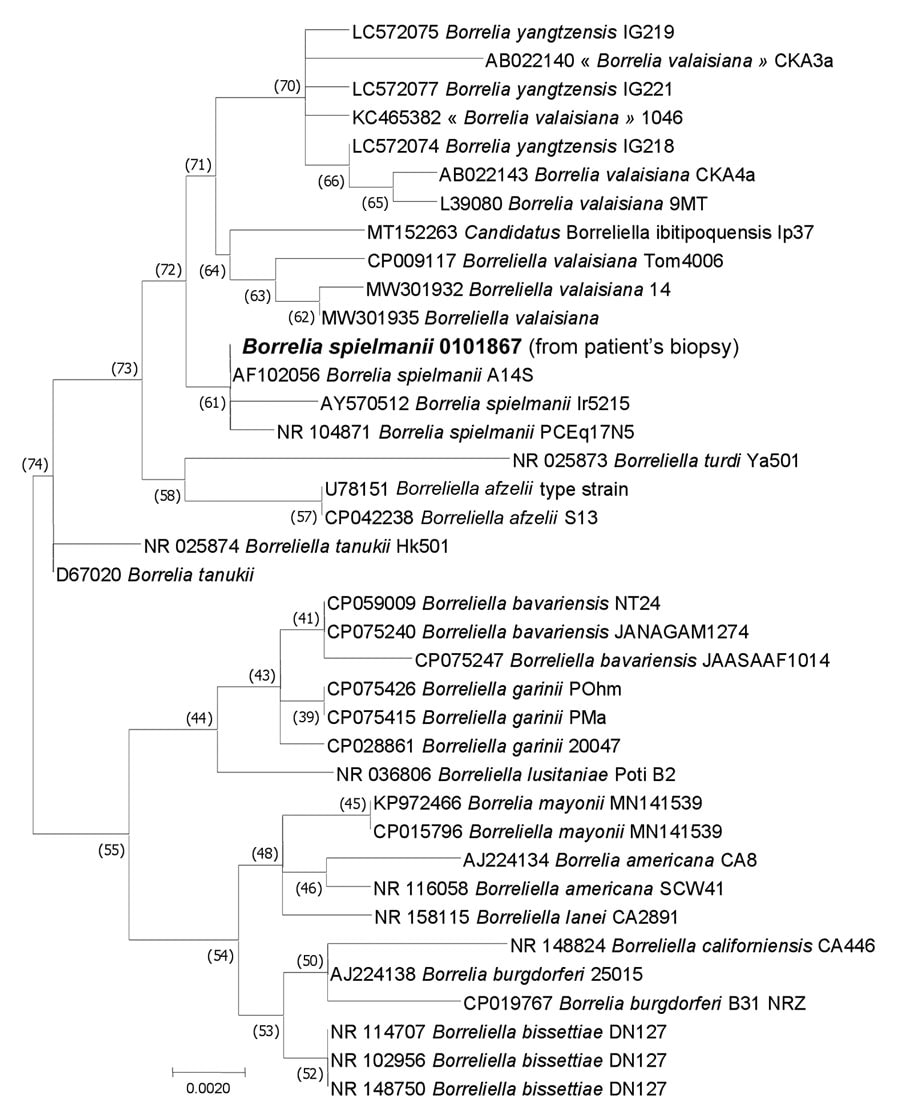
Figure 2. Maximum-likelihood phylogenetic tree of the 16s rRNA gene (rrs) of Borrelia genus bacteria showing the position of the B. spielmanii sequence obtained from the patient (large bold font) Evolutionary analyses were conducted using TOPALi version 2.5 (http://www.topali.org). The sequences of the 16S rDNA amplified in this study with other 12S rDNA tick sequences available on GenBank (910 positions in the final dataset) were aligned using ClustalW (https://www.genome.jp/tools-bin/clustalw) implemented on BioEdit version 3 (https://bioedit.software.informer.com). The evolutionary history was inferred by using the maximum likelihood method based on the Hasegawa–Kishino–Yano model plus invariate sites plus gamma distribution. The percentage of trees in which the associated taxa clustered together is shown next to the branches. GenBank accession numbers are provided. Scale bar indicates nucleotide sequence divergence.
Main Article
Page created: September 20, 2023
Page updated: October 23, 2023
Page reviewed: October 23, 2023
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.