 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
| ||||||||||
|
|
|
|
|
Persons using assistive technology might not be able to fully access information in this file. For assistance, please send e-mail to: mmwrq@cdc.gov. Type 508 Accommodation and the title of the report in the subject line of e-mail. Treatment of TuberculosisAmerican Thoracic Society, CDC, and Infectious Diseases Society of AmericaAn erratum has been published for this article. To view the erratum, please click here. This Official Joint Statement of the American Thoracic Society, CDC, and the Infectious Diseases Society of America was approved by the ATS Board of Directors, by CDC, and by the Council of the IDSA in October 2002. This report appeared in the American Journal of Respiratory and Critical Care Medicine (2003;167:603--62) and is being reprinted as a courtesy to the American Thoracic Society, the Infectious Diseases Society of America, and the MMWR readership. PurposeThe recommendations in this document are intended to guide the treatment of tuberculosis in settings where mycobacterial cultures, drug susceptibility testing, radiographic facilities, and second-line drugs are routinely available. In areas where these resources are not available, the recommendations provided by the World Health Organization, the International Union against Tuberculosis, or national tuberculosis control programs should be followed. What's New In This Document
SummaryResponsibility for Successful Treatment The overall goals for treatment of tuberculosis are 1) to cure the individual patient, and 2) to minimize the transmission of Mycobacterium tuberculosis to other persons. Thus, successful treatment of tuberculosis has benefits both for the individual patient and the community in which the patient resides. For this reason the prescribing physician, be he/she in the public or private sector, is carrying out a public health function with responsibility not only for prescribing an appropriate regimen but also for successful completion of therapy. Prescribing physician responsibility for treatment completion is a fundamental principle in tuberculosis control. However, given a clear understanding of roles and responsibilities, oversight of treatment may be shared between a public health program and a private physician. Organization and Supervision of Treatment Treatment of patients with tuberculosis is most successful within a comprehensive framework that addresses both clinical and social issues of relevance to the patient. It is essential that treatment be tailored and supervision be based on each patient's clinical and social circumstances (patient-centered care). Patients may be managed in the private sector, by public health departments, or jointly, but in all cases the health department is ultimately responsible for ensuring that adequate, appropriate diagnostic and treatment services are available, and for monitoring the results of therapy. It is strongly recommended that patient-centered care be the initial management strategy, regardless of the source of supervision. This strategy should always include an adherence plan that emphasizes directly observed therapy (DOT), in which patients are observed to ingest each dose of antituberculosis medications, to maximize the likelihood of completion of therapy. Programs utilizing DOT as the central element in a comprehensive, patient-centered approach to case management (enhanced DOT) have higher rates of treatment completion than less intensive strategies. Each patient's management plan should be individualized to incorporate measures that facilitate adherence to the drug regimen. Such measures may include, for example, social service support, treatment incentives and enablers, housing assistance, referral for treatment of substance abuse, and coordination of tuberculosis services with those of other providers. Recommended Treatment Regimens The recommended treatment regimens are, in large part, based on evidence from clinical trials and are rated on the basis of a system developed by the United States Public Health Service (USPHS) and the Infectious Diseases Society of America (IDSA). The rating system includes a letter (A, B, C, D, or E) that indicates the strength of the recommendation and a roman numeral (I, II, or III) that indicates the quality of evidence supporting the recommendation (Table 1). There are four recommended regimens for treating patients with tuberculosis caused by drug-susceptible organisms. Although these regimens are broadly applicable, there are modifications that should be made under specified circumstances, described subsequently. Each regimen has an initial phase of 2 months followed by a choice of several options for the continuation phase of either 4 or 7 months. The recommended regimens together with the number of doses specified by the regimen are described in Table 2. The initial phases are denoted by a number (1, 2, 3, or 4) and the continuation phases that relate to the initial phase are denoted by the number plus a letter designation (a, b, or c). Drug doses are shown in Tables 3, 4, and 5. The general approach to treatment is summarized in Figure 1. Because of the relatively high proportion of adult patients with tuberculosis caused by organisms that are resistant to isoniazid, four drugs are necessary in the initial phase for the 6-month regimen to be maximally effective. Thus, in most circumstances, the treatment regimen for all adults with previously untreated tuberculosis should consist of a 2-month initial phase of isoniazid (INH), rifampin (RIF), pyrazinamide (PZA), and ethambutol (EMB) (Table 2, Regimens 1--3). If (when) drug susceptibility test results are known and the organisms are fully susceptible, EMB need not be included. For children whose visual acuity cannot be monitored, EMB is usually not recommended except when there is an increased likelihood of the disease being caused by INH-resistant organisms (Table 6) or when the child has "adult-type" (upper lobe infiltration, cavity formation) tuberculosis. If PZA cannot be included in the initial phase of treatment, or if the isolate is resistant to PZA alone (an unusual circumstance), the initial phase should consist of INH, RIF, and EMB given daily for 2 months (Regimen 4). Examples of circumstances in which PZA may be withheld include severe liver disease, gout, and, perhaps, pregnancy. EMB should be included in the initial phase of Regimen 4 until drug susceptibility is determined. The initial phase may be given daily throughout (Regimens 1 and 4), daily for 2 weeks and then twice weekly for 6 weeks (Regimen 2), or three times weekly throughout (Regimen 3). For patients receiving daily therapy, EMB can be discontinued as soon as the results of drug susceptibility studies demonstrate that the isolate is susceptible to INH and RIF. When the patient is receiving less than daily drug administration, expert opinion suggests that EMB can be discontinued safely in less than 2 months (i.e., when susceptibility test results are known), but there is no evidence to support this approach. Although clinical trials have shown that the efficacy of streptomycin (SM) is approximately equal to that of EMB in the initial phase of treatment, the increasing frequency of resistance to SM globally has made the drug less useful. Thus, SM is not recommended as being interchangeable with EMB unless the organism is known to be susceptible to the drug or the patient is from a population in which SM resistance is unlikely. The continuation phase (Table 2) of treatment is given for either 4 or 7 months. The 4-month continuation phase should be used in the large majority of patients. The 7-month continuation phase is recommended only for three groups: patients with cavitary pulmonary tuberculosis caused by drug-susceptible organisms and whose sputum culture obtained at the time of completion of 2 months of treatment is positive; patients whose initial phase of treatment did not include PZA; and patients being treated with once weekly INH and rifapentine and whose sputum culture obtained at the time of completion of the initial phase is positive. The continuation phase may be given daily (Regimens 1a and 4a), two times weekly by DOT (Regimens 1b, 2a, and 4b), or three times weekly by DOT (Regimen 3a). For human immunodeficiency virus (HIV)-seronegative patients with noncavitary pulmonary tuberculosis (as determined by standard chest radiography), and negative sputum smears at completion of 2 months of treatment, the continuation phase may consist of rifapentine and INH given once weekly for 4 months by DOT (Regimens 1c and 2b) (Figure 1). If the culture at completion of the initial phase of treatment is positive, the once weekly INH and rifapentine continuation phase should be extended to 7 months. All of the 6-month regimens, except the INH--rifapentine once weekly continuation phase for persons with HIV infection (Rating EI), are rated as AI or AII, or BI or BII, in both HIV-infected and uninfected patients. The once-weekly continuation phase is contraindicated (Rating EI) in patients with HIV infection because of an unacceptable rate of failure/relapse, often with rifamycin-resistant organisms. For the same reason twice weekly treatment, either as part of the initial phase (Regimen 2) or continuation phase (Regimens 1b and 2a), is not recommended for HIV-infected patients with CD4+ cell counts <100 cells/µl. These patients should receive either daily (initial phase) or three times weekly (continuation phase) treatment. Regimen 4 (and 4a/4b), a 9-month regimen, is rated CI for patients without HIV infection and CII for those with HIV infection. Deciding To Initiate Treatment The decision to initiate combination antituberculosis chemotherapy should be based on epidemiologic information; clinical, pathological, and radiographic findings; and the results of microscopic examination of acid-fast bacilli (AFB)--stained sputum (smears) (as well as other appropriately collected diagnostic specimens) and cultures for mycobacteria. A purified protein derivative (PPD)-tuberculin skin test may be done at the time of initial evaluation, but a negative PPD-tuberculin skin test does not exclude the diagnosis of active tuberculosis. However, a positive PPD-tuberculin skin test supports the diagnosis of culture-negative pulmonary tuberculosis, as well as latent tuberculosis infection in persons with stable abnormal chest radiographs consistent with inactive tuberculosis (see below). If the suspicion of tuberculosis is high or the patient is seriously ill with a disorder, either pulmonary or extrapulmonary, that is thought possibly to be tuberculosis, combination chemotherapy using one of the recommended regimens should be initiated promptly, often before AFB smear results are known and usually before mycobacterial culture results have been obtained. A positive AFB smear provides strong inferential evidence for the diagnosis of tuberculosis. If the diagnosis is confirmed by isolation of M. tuberculosis or a positive nucleic acid amplification test, treatment can be continued to complete a standard course of therapy (Figure 1). When the initial AFB smears and cultures are negative, a diagnosis other than tuberculosis should be considered and appropriate evaluations undertaken. If no other diagnosis is established and the PPD-tuberculin skin test is positive (in this circumstance a reaction of 5 mm or greater induration is considered positive), empirical combination chemotherapy should be initiated. If there is a clinical or radiographic response within 2 months of initiation of therapy and no other diagnosis has been established, a diagnosis of culture-negative pulmonary tuberculosis can be made and treatment continued with an additional 2 months of INH and RIF to complete a total of 4 months of treatment, an adequate regimen for culture-negative pulmonary tuberculosis (Figure 2). If there is no clinical or radiographic response by 2 months, treatment can be stopped and other diagnoses including inactive tuberculosis considered. If AFB smears are negative and suspicion for active tuberculosis is low, treatment can be deferred until the results of mycobacterial cultures are known and a comparison chest radiograph is available (usually within 2 months) (Figure 2). In low-suspicion patients not initially being treated, if cultures are negative, the PPD-tuberculin skin test is positive (5 mm or greater induration), and the chest radiograph is unchanged after 2 months, one of the three regimens recommended for the treatment of latent tuberculosis infection could be used. These include (1) INH for a total of 9 months, (2) RIF with or without INH for a total of 4 months, or (3) RIF and PZA for a total of 2 months. Because of reports of an increased rate of hepatotoxicity with the RIF--PZA regimen, it should be reserved for patients who are not likely to complete a longer course of treatment, can be monitored closely, and do not have contraindications to the use of this egimen. Baseline and Follow-Up Evaluations Patients suspected of having tuberculosis should have appropriate specimens collected for microscopic examination and mycobacterial culture. When the lung is the site of disease, three sputum specimens should be obtained. Sputum induction with hypertonic saline may be necessary to obtain specimens and bronchoscopy (both performed under appropriate infection control measures) may be considered for patients who are unable to produce sputum, depending on the clinical circumstances. Susceptibility testing for INH, RIF, and EMB should be performed on a positive initial culture, regardless of the source of the specimen. Second-line drug susceptibility testing should be done only in reference laboratories and be limited to specimens from patients who have had prior therapy, who are contacts of patients with drug-resistant tuberculosis, who have demonstrated resistance to rifampin or to other first-line drugs, or who have positive cultures after more than 3 months of treatment. It is recommended that all patients with tuberculosis have counseling and testing for HIV infection, at least by the time treatment is initiated, if not earlier. For patients with HIV infection, a CD4+ lymphocyte count should be obtained. Patients with risk factors for hepatitis B or C viruses (e.g., injection drug use, foreign birth in Asia or Africa, HIV infection) should have serologic tests for these viruses. For all adult patients baseline measurements of serum amino transferases (aspartate aminotransferase [AST], alanine aminotransferase [ALT]), bilirubin, alkaline phosphatase, and serum creatinine and a platelet count should be obtained. Testing of visual acuity and red-green color discrimination should be obtained when EMB is to be used. During treatment of patients with pulmonary tuberculosis, a sputum specimen for microscopic examination and culture should be obtained at a minimum of monthly intervals until two consecutive specimens are negative on culture. More frequent AFB smears may be useful to assess the early response to treatment and to provide an indication of infectiousness. For patients with extrapulmonary tuberculosis the frequency and kinds of evaluations will depend on the site involved. In addition, it is critical that patients have clinical evaluations at least monthly to identify possible adverse effects of the antituberculosis medications and to assess adherence. Generally, patients do not require follow-up after completion of therapy but should be instructed to seek care promptly if signs or symptoms recur. Routine measurements of hepatic and renal function and platelet count are not necessary during treatment unless patients have baseline abnormalities or are at increased risk of hepatotoxicity (e.g., hepatitis B or C virus infection, alcohol abuse). At each monthly visit patients taking EMB should be questioned regarding possible visual disturbances including blurred vision or scotomata; monthly testing of visual acuity and color discrimination is recommended for patients taking doses that on a milligram per kilogram basis are greater than those listed in Table 5 and for patients receiving the drug for longer than 2 months. Identification and Management of Patients at Increased Risk of Treatment Failure and Relapse The presence of cavitation on the initial chest radiograph combined with having a positive sputum culture at the time the initial phase of treatment is completed has been shown in clinical trials to identify patients at high risk for adverse outcomes (treatment failure, usually defined by positive cultures after 4 months of treatment, or relapse, defined by recurrent tuberculosis at any time after completion of treatment and apparent cure). For this reason it is particularly important to conduct a microbiological evaluation 2 months after initiation of treatment (Figure 1). Approximately 80% of patients with pulmonary tuberculosis caused by drug-susceptible organisms who are started on standard four-drug therapy will have negative sputum cultures at this time. Patients with positive cultures after 2 months of treatment should undergo careful evaluation to determine the cause. For patients who have positive cultures after 2 months of treatment and have not been receiving DOT, the most common reason is nonadherence to the regimen. Other possibilities, especially for patients receiving DOT, include extensive cavitary disease at the time of diagnosis, drug resistance, malabsorption of drugs, laboratory error, and biological variation in response. In USPHS Study 22, nearly 21% of patients in the control arm of the study (a continuation phase of twice weekly INH and RIF) who had both cavitation on the initial chest radiograph and a positive culture at the 2-month juncture relapsed. Patients who had only one of these factors (either cavitation or a positive 2-month culture) had relapse rates of 5--6% compared with 2% for patients who had neither risk factor. In view of this evidence, it is recommended that, for patients who have cavitation on the initial chest radiograph and whose 2-month culture is positive, the minimum duration of treatment should be 9 months (a total of 84--273 doses depending on whether the drugs are given daily or intermittently) (Figure 1 and Table 2). The recommendation to lengthen the continuation phase of treatment is based on expert opinion and on the results of a study of the optimal treatment duration for patients with silicotuberculosis showing that extending treatment from 6 to 8 months greatly reduced the rate of relapse (Rating AIII). The recommendation is also supported by the results of a trial in which the once weekly INH--rifapentine continuation phase was extended to 7 months for patients at high risk of relapse. The rate of relapse was reduced significantly compared with historical control subjects from another trial in which the continuation phase was 4 months. For patients who have either cavitation on the initial film or a positive culture after completing the initial phase of treatment (i.e., at 2 months), the rates of relapse were 5--6%. In this group decisions to prolong the continuation phase should be made on an individual basis. Completion of Treatment A full course of therapy (completion of treatment) is determined more accurately by the total number of doses taken, not solely by the duration of therapy. For example, the "6-month" daily regimen (given 7 days/week; see below) should consist of at least 182 doses of INH and RIF, and 56 doses of PZA. Thus, 6 months is the minimum duration of treatment and accurately indicates the amount of time the drugs are given only if there are no interruptions in drug administration. In some cases, either because of drug toxicity or nonadherence to the treatment regimen, the specified number of doses cannot be administered within the targeted period. In such cases the goal is to deliver the specified number of doses within a recommended maximum time. For example, for a 6-month daily regimen the 182 doses should be administered within 9 months of beginning treatment. If treatment is not completed within this period, the patient should be assessed to determine the appropriate action to take---continuing treatment for a longer duration or restarting treatment from the beginning, either of which may require more restrictive measures to be used to ensure completion. Clinical experience suggests that patients being managed by DOT administered 5 days/week have a rate of successful therapy equivalent to those being given drugs 7 days/week. Thus, "daily therapy" may be interpreted to mean DOT given 5 days/week and the required number of doses adjusted accordingly. For example, for the 6-month "daily" regimen given 5 days/week the planned total number of doses is 130. (Direct observation of treatment given 5 days/week has been used in a number of clinical trials, including USPHS Study 22, but has not been evaluated in a controlled trial; thus, this modification should be rated AIII.) As an option, patients might be given the medications to take without DOT on weekends. Interruptions in treatment may have a significant effect on the duration of therapy. Reinstitution of treatment must take into account the bacillary load of the patient, the point in time when the interruption occurred, and the duration of the interruption. In general, the earlier in treatment and the longer the duration of the interruption, the more serious the effect and the greater the need to restart therapy from the beginning. Practical Aspects of Patient Management During Treatment The first-line antituberculosis medications should be administered together; split dosing should be avoided. Fixed-dose combination preparations may be administered more easily than single drug tablets and may decrease the risk of acquired drug resistance and medication errors. Fixed-dose combinations may be used when DOT is given daily and are especially useful when DOT is not possible, but they are not formulated for use with intermittent dosing. It should be noted that for patients weighing more than 90 kg the dose of PZA in the three-drug combination is insufficient and additional PZA tablets are necessary. There are two combination formulations approved for use in the United States: INH and RIF (Rifamate®) and INH, RIF, and PZA (Rifater®). Providers treating patients with tuberculosis must be especially vigilant for drug interactions. Given the frequency of comorbid conditions, it is quite common for patients with tuberculosis to be taking a variety of other medications, the effects of which may be altered by the antituberculosis medications, especially the rifamycins. These interactions are described in Section 7, Drug Interactions. Adverse effects, especially gastrointestinal upset, are relatively common in the first few weeks of antituberculosis therapy; however, first-line antituberculosis drugs, particularly RIF, must not be discontinued because of minor side effects. Although ingestion with food delays or moderately decreases the absorption of antituberculosis drugs, the effects of food are of little clinical significance. Thus, if patients have epigastric distress or nausea with the first-line drugs, dosing with meals or changing the hour of dosing is recommended. Administration with food is preferable to splitting a dose or changing to a second-line drug. Drug-induced hepatitis, the most serious common adverse effect, is defined as a serum AST level more than three times the upper limit of normal in the presence of symptoms, or more than five times the upper limit of normal in the absence of symptoms. If hepatitis occurs INH, RIF, and PZA, all potential causes of hepatic injury, should be stopped immediately. Serologic testing for hepatitis viruses A, B, and C (if not done at baseline) should be performed and the patient questioned carefully regarding exposure to other possible hepatotoxins, especially alcohol. Two or more antituberculosis medications without hepatotoxicity, such as EMB, SM, amikacin/kanamycin, capreomycin, or a fluoroquinolone (levofloxacin, moxifloxacin, or gatifloxacin), may be used until the cause of the hepatitis is identified. Once the AST level decreases to less than two times the upper limit of normal and symptoms have significantly improved, the first-line medications should be restarted in sequential fashion. Close monitoring, with repeat measurements of serum AST and bilirubin and symptom review, is essential in managing these patients. Treatment in Special Situations HIV infection Recommendations for the treatment of tuberculosis in HIV-infected adults are, with a few exceptions, the same as those for HIV-uninfected adults (Table 2). The INH--rifapentine once weekly continuation phase (Regimens 1c and 2b) is contraindicated in HIV-infected patients because of an unacceptably high rate of relapse, frequently with organisms that have acquired resistance to rifamycins. The development of acquired rifampin resistance has also been noted among HIV-infected patients with advanced immunosuppression treated with twice weekly rifampin- or rifabutin-based regimens. Consequently, patients with CD4+ cell counts <100/µl should receive daily or three times weekly treatment (Regimen 1/1a or Regimen 3/3a). DOT and other adherence-promoting strategies are especially important for patients with HIV-related tuberculosis. Management of HIV-related tuberculosis is complex and requires expertise in the management of both HIV disease and tuberculosis. Because HIV-infected patients are often taking numerous medications, some of which interact with antituberculosis medications, it is strongly encouraged that experts in the treatment of HIV-related tuberculosis be consulted. A particular concern is the interaction of rifamycins with antiretroviral agents and other antiinfective drugs. Rifampin can be used for the treatment of tuberculosis with certain combinations of antiretroviral agents. Rifabutin, which has fewer problematic drug interactions, may also be used in place of rifampin and appears to be equally effective although the doses of rifabutin and antiretroviral agents may require adjustment. As new antiretroviral agents and more pharmacokinetic data become available, these recommendations are likely to be modified. On occasion, patients with HIV-related tuberculosis may experience a temporary exacerbation of symptoms, signs, or radiographic manifestations of tuberculosis while receiving antituberculosis treatment. This clinical or radiographic worsening (paradoxical reaction) occurs in HIV-infected patients with active tuberculosis and is thought to be the result of immune reconstitution as a consequence of effective antiretroviral therapy. Symptoms and signs may include high fevers, lymphadenopathy, expanding central nervous system lesions, and worsening of chest radiographic findings. The diagnosis of a paradoxical reaction should be made only after a thorough evaluation has excluded other etiologies, particularly tuberculosis treatment failure. Nonsteroidal antiinflammatory agents may be useful for symptomatic relief. For severe paradoxical reactions, prednisone (1--2 mg/kg per day for 1--2 weeks, then in gradually decreasing doses) may be used, although there are no data from controlled trials to support this approach (Rating CIII). Children Because of the high risk of disseminated tuberculosis in infants and children younger than 4 years of age, treatment should be started as soon as the diagnosis of tuberculosis is suspected. In general, the regimens recommended for adults are also the regimens of choice for infants, children, and adolescents with tuberculosis, with the exception that ethambutol is not used routinely in children. Because there is a lower bacillary burden in childhood-type tuberculosis there is less concern with the development of acquired drug resistance. However, children and adolescents may develop "adult-type" tuberculosis with upper lobe infiltration, cavitation, and sputum production. In such situations an initial phase of four drugs should be given until susceptibility is proven. When clinical or epidemiologic circumstances (Table 6) suggest an increased probability of INH resistance, EMB can be used safely at a dose of 15--20 mg/kg per day, even in children too young for routine eye testing. Streptomycin, kanamycin, or amikacin also can be used as the fourth drug, when necessary. Most studies of treatment in children have used 6 months of INH and RIF supplemented during the first 2 months with PZA. This three-drug combination has a success rate of greater than 95% and an adverse drug reaction rate of less than 2%. Most treatment studies of intermittent dosing in children have used daily drug administration for the first 2 weeks to 2 months. DOT should always be used in treating children. Because it is difficult to isolate M. tuberculosis from a child with pulmonary tuberculosis, it is frequently necessary to rely on the results of drug susceptibility tests of the organisms isolated from the presumed source case to guide the choice of drugs for the child. In cases of suspected drug-resistant tuberculosis in a child or when a source case isolate is not available, specimens for microbiological evaluation should be obtained via early morning gastric aspiration, bronchoalveolar lavage, or biopsy. In general, extrapulmonary tuberculosis in children can be treated with the same regimens as pulmonary disease. Exceptions are disseminated tuberculosis and tuberculous meningitis, for which there are inadequate data to support 6-month therapy; thus 9--12 months of treatment is recommended. The optimal treatment of pulmonary tuberculosis in children and adolescents with HIV infection is unknown. The American Academy of Pediatrics recommends that initial therapy should always include at least three drugs, and the total duration of therapy should be at least 9 months, although there are no data to support this recommendation. Extrapulmonary tuberculosis The basic principles that underlie the treatment of pulmonary tuberculosis also apply to extrapulmonary forms of the disease. Although relatively few studies have examined treatment of extrapulmonary tuberculosis, increasing evidence suggests that 6- to 9-month regimens that include INH and RIF are effective. Thus, a 6-month course of therapy is recommended for treating tuberculosis involving any site with the exception of the meninges, for which a 9- 12-month regimen is recommended. Prolongation of therapy also should be considered for patients with tuberculosis in any site that is slow to respond. The addition of corticosteroids is recommended for patients with tuberculous pericarditis and tuberculous meningitis. Culture-negative pulmonary tuberculosis and radiographic evidence of prior pulmonary tuberculosis Failure to isolate M. tuberculosis from persons suspected of having pulmonary tuberculosis on the basis of clinical features and chest radiographic examination does not exclude a diagnosis of active tuberculosis. Alternative diagnoses should be considered carefully and further appropriate diagnostic studies undertaken in persons with apparent culture-negative tuberculosis. The general approach to management is shown in Figure 2. A diagnosis of tuberculosis can be strongly inferred by the clinical and radiographic response to antituberculosis treatment. Careful reevaluation should be performed after 2 months of therapy to determine whether there has been a response attributable to antituberculosis treatment. If either clinical or radiographic improvement is noted and no other etiology is identified, treatment should be continued for active tuberculosis. Treatment regimens in this circumstance include one of the standard 6-month chemotherapy regimens or INH, RIF, PZA, and EMB for 2 months followed by INH and RIF for an additional 2 months (4 months total). However, HIV-infected patients with culture-negative pulmonary tuberculosis should be treated for a minimum of 6 months. Persons with a positive tuberculin skin test who have radiographic evidence of prior tuberculosis (e.g., upper lobe fibronodular infiltrations) but who have not received adequate therapy are at increased risk for the subsequent development of tuberculosis. Unless previous radiographs are available showing that the abnormality is stable, it is recommended that sputum examination (using sputum induction if necessary) be performed to assess the possibility of active tuberculosis being present. Also, if the patient has symptoms of tuberculosis related to an extrapulmonary site, an appropriate evaluation should be undertaken. Once active tuberculosis has been excluded (i.e., by negative cultures and a stable chest radiograph), the treatment regimens are those used for latent tuberculosis infection: INH for 9 months, RIF (with or without INH) for 4 months, or RIF and PZA for 2 months (for patients who are unlikely to complete a longer course and who can be monitored closely) (Figure 2). Renal insufficiency and end-stage renal disease Specific dosing guidelines for patients with renal insufficiency and end-stage renal disease are provided in Table 15. For patients undergoing hemodialysis, administration of all drugs after dialysis is preferred to facilitate DOT and to avoid premature removal of drugs such as PZA and cycloserine. To avoid toxicity it is important to monitor serum drug concentrations in persons with renal failure who are taking cycloserine or EMB. There is little information concerning the effects of peritoneal dialysis on clearance of antituberculosis drugs. Liver disease INH, RIF, and PZA all can cause hepatitis that may result in additional liver damage in patients with preexisting liver disease. However, because of the effectiveness of these drugs (particularly INH and RIF), they should be used if at all possible, even in the presence of preexisting liver disease. If serum AST is more than three times normal before the initiation of treatment (and the abnormalities are not thought to be caused by tuberculosis), several treatment options exist. One option is to treat with RIF, EMB, and PZA for 6 months, avoiding INH. A second option is to treat with INH and RIF for 9 months, supplemented by EMB until INH and RIF susceptibility are demonstrated, thereby avoiding PZA. For patients with severe liver disease a regimen with only one hepatotoxic agent, generally RIF plus EMB, could be given for 12 months, preferably with another agent, such as a fluoroquinolone, for the first 2 months; however, there are no data to support this recommendation. In all patients with preexisting liver disease, frequent clinical and laboratory monitoring should be performed to detect drug-induced hepatic injury. Pregnancy and breastfeeding Because of the risk of tuberculosis to the fetus, treatment of tuberculosis in pregnant women should be initiated whenever the probability of maternal disease is moderate to high. The initial treatment regimen should consist of INH, RIF, and EMB. Although all of these drugs cross the placenta, they do not appear to have teratogenic effects. Streptomycin is the only antituberculosis drug documented to have harmful effects on the human fetus (congenital deafness) and should not be used. Although detailed teratogenicity data are not available, PZA can probably be used safely during pregnancy and is recommended by the World Health Organization (WHO) and the International Union against Tuberculosis and Lung Disease (IUATLD). If PZA is not included in the initial treatment regimen, the minimum duration of therapy is 9 months. Breastfeeding should not be discouraged for women being treated with the first-line antituberculosis agents because the small concentrations of these drugs in breast milk do not produce toxicity in the nursing newborn. Conversely, drugs in breast milk should not be considered to serve as effective treatment for tuberculosis or for latent tuberculosis infection in a nursing infant. Pyridoxine supplementation (25 mg/day) is recommended for all women taking INH who are either pregnant or breastfeeding. The amount of pyridoxine in multivitamins is variable but generally less than the needed amount. Management of Relapse, Treatment Failure, and Drug Resistance Relapse refers to the circumstance in which a patient becomes and remains culture negative while receiving therapy but, at some point after completion of therapy, either becomes culture positive again or has clinical or radiographic deterioration that is consistent with active tuberculosis. In the latter situation rigorous efforts should be made to establish a diagnosis and to obtain microbiological confirmation of the relapse to enable testing for drug resistance. Most relapses occur within the first 6--12 months after completion of therapy. In nearly all patients with tuberculosis caused by drug-susceptible organisms and who were treated with rifamycin-containing regimens using DOT, relapses occur with susceptible organisms. However, in patients who received self-administered therapy or a nonrifamycin regimen and who have a relapse, the risk of acquired drug resistance is substantial. In addition, if initial drug susceptibility testing was not performed and the patient fails or relapses with a rifamycin-containing regimen given by DOT, there is a high likelihood that the organisms were resistant from the outset. The selection of empirical treatment for patients with relapse should be based on the prior treatment scheme and severity of disease. For patients with tuberculosis that was caused by drug-susceptible organisms and who were treated under DOT, initiation of the standard four-drug regimen is appropriate until the results of drug susceptibility tests are available. However, for patients who have life-threatening forms of tuberculosis, at least three additional agents to which the organisms are likely to be susceptible should be included. For patients with relapse who did not receive DOT, who were not treated with a rifamycin-based regimen, or who are known or presumed to have had irregular treatment, it is prudent to infer that drug resistance is present and to begin an expanded regimen with INH, RIF, and PZA plus an additional two or three agents based on the probability of in vitro susceptibility. Usual agents to be employed would include a fluoroquinolone (levofloxacin, moxifloxacin, or gatifloxacin), an injectable agent such as SM (if not used previously and susceptibility to SM had been established), amikacin, kanamycin, or capreomycin, with or without an additional oral drug. Treatment failure is defined as continued or recurrently positive cultures during the course of antituberculosis therapy. After 3 months of multidrug therapy for pulmonary tuberculosis caused by drug-susceptible organisms, 90--95% of patients will have negative cultures and show clinical improvement. Thus, patients with positive cultures after 3 months of what should be effective treatment must be evaluated carefully to identify the cause of the delayed conversion. Patients whose sputum cultures remain positive after 4 months of treatment should be deemed treatment failures. Possible reasons for treatment failure in patients receiving appropriate regimens include nonadherence to the drug regimen (the most common reason), drug resistance, malabsorption of drugs, laboratory error, and extreme biological variation in response. If treatment failure occurs, early consultation with a specialty center is strongly advised. If failure is likely due to drug resistance and the patient is not seriously ill, an empirical retreatment regimen could be started or administration of an altered regimen could be deferred until results of drug susceptibility testing from a recent isolate are available. If the patient is seriously ill or sputum AFB smears are positive, an empirical regimen should be started immediately and continued until susceptibility tests are available. For patients who have treatment failure, M. tuberculosis isolates should be sent promptly to a reference laboratory for drug susceptibility testing to both first- and second-line agents. A fundamental principle in managing patients with treatment failure is never to add a single drug to a failing regimen; so doing leads to acquired resistance to the new drug. Instead, at least two, and preferably three, new drugs to which susceptibility could logically be inferred should be added to lessen the probability of further acquired resistance. Empirical retreatment regimens might include a fluoroquinolone, an injectable agent such as SM (if not used previously and the patient is not from an area of the world having high rates of SM resistance), amikacin, kanamycin, or capreomycin, and an additional oral agent such as p-aminosalicylic acid (PAS), cycloserine, or ethionamide. Once drug-susceptibility test results are available, the regimen should be adjusted according to the results. Patients having tuberculosis caused by strains of M. tuberculosis resistant to at least INH and RIF (multidrug-resistant [MDR]) are at high risk for treatment failure and further acquired drug resistance. Such patients should be referred to or consultation obtained from specialized treatment centers as identified by the local or state health departments or CDC. Although patients with strains resistant to RIF alone have a better prognosis than patients with MDR strains, they are also at increased risk for treatment failure and additional resistance and should be managed in consultation with an expert. Definitive randomized or controlled studies have not been performed to establish optimum regimens for treating patients with the various patterns of drug-resistant tuberculosis; thus, treatment recommendations are based on expert opinion, guided by a set of general principles specified in Section 9, Management of Relapse, Treatment Failure, and Drug Resistance. Table 16 contains treatment regimens suggested for use in patients with various patterns of drug-resistant tuberculosis (all are rated AIII). The role of resectional surgery in the management of patients with extensive pulmonary MDR tuberculosis has not been established in randomized studies and results have been mixed. Surgery should be performed by surgeons with experience in these situations and only after the patient has received several months of intensive chemotherapy. Expert opinion suggests that chemotherapy should be continued for 1--2 years postoperatively to prevent relapse. Treatment of Tuberculosis in Low-Income Countries: Recommendations of the WHO and Guidelines from the IUATLD To place the current guidelines in an international context it is necessary to have an understanding of the approaches to treatment of tuberculosis in high-incidence, low-income countries. It is important to recognize that the American Thoracic Society/CDC/Infectious Diseases Society of America (ATS/CDC/IDSA) recommendations cannot be assumed to be applicable under all epidemiologic and economic circumstances. The incidence of tuberculosis and the resources with which to confront the disease to an important extent determine the approaches used. Given the increasing proportion of patients in low-incidence countries who were born in high-incidence countries, it is also important for persons managing these cases to be familiar with the approaches used in the countries of origin. The major international recommendations and guidelines for treating tuberculosis are those of the WHO and of the IUATLD. The WHO document was developed by an expert committee whereas the IUATLD document is a distillation of IUATLD practice, validated in the field. The WHO and IUATLD documents target, in general, countries in which mycobacterial culture, drug susceptibility testing, radiographic facilities, and second-line drugs are not widely available as a routine. A number of differences exist between these new ATS/CDC/IDSA recommendations, and the current tuberculosis treatment recommendations of the WHO and guidelines of the IUATLD. Both international sets of recommendations are built around a national case management strategy called "DOTS," the acronym for "directly observed therapy, short course," in which direct observation of therapy (DOT) is only one of five key elements. The five components of DOTS are 1) government commitment to sustained tuberculosis control activities, 2) case detection by sputum smear microscopy among symptomatic patients self-reporting to health services, 3) a standardized treatment regimen of 6--8 months for at least all confirmed sputum smear--positive cases, with DOT for at least the initial 2 months, 4) a regular, uninterrupted supply of all essential antituberculosis drugs, and 5) a standardized recording and reporting system that enables assessment of treatment results for each patient and of the tuberculosis control program overall. A number of other differences exist as well:
A Research Agenda for Tuberculosis Treatment New antituberculosis drugs are needed for three main reasons: 1) to shorten or otherwise simplify treatment of tuberculosis caused by drug-susceptible organisms, 2) to improve treatment of drug-resistant tuberculosis, and 3) to provide more efficient and effective treatment of latent tuberculosis infection. No truly novel compounds that are likely to have a significant impact on tuberculosis treatment are close to clinical trials. However, further work to optimize the effectiveness of once-a-week rifapentine regimens using higher doses of the drug and using rifapentine in combination with moxifloxacin is warranted, on the basis of experimental data. New categories of drugs that have shown promise for use in treating tuberculosis include the nitroimidazopyrans and the oxazolidinones. Experimental data also suggest that a drug to inhibit an enzyme, isocitrate lyase, thought to be necessary for maintaining the latent state, might be useful for treatment of latent tuberculosis infection. A number of other interventions that might lead to improved treatment outcome have been suggested, although none has undergone rigorous clinical testing. These include various drug delivery systems, cytokine inhibitors, administration of "protective" cytokines such as interferon-g and interleukin-2, and nutritional supplements, especially vitamin A and zinc. Research is also needed to identify factors that are predictive of a greater or lesser risk of relapse to determine optimal length of treatment. Identification of such factors would enable more efficient targeting of resources to supervise treatment. In addition, identification of behavioral factors that identify patients at greater or lesser likelihood of being adherent to therapy would also enable more efficient use of DOT. 1. Introduction and BackgroundSince 1971 the American Thoracic Society (ATS) and CDC have regularly collaborated to develop joint guidelines for the diagnosis, treatment, prevention, and control of tuberculosis (1). These documents have been intended to guide both public health programs and health care providers in all aspects of the clinical and public health management of tuberculosis in low-incidence countries, with a particular focus on the United States. The most recent version of guidelines for the treatment of tuberculosis was published in 1994 (2). The current document differs from its predecessor in a number of important areas that are summarized above. The process by which this revision of the recommendations for treatment was developed was modified substantially from the previous versions. For the first time the Infectious Diseases Society of America (IDSA) has become a cosponsor of the statement, together with the ATS and CDC. The IDSA has had representation on prior statement committees but has not previously been a cosponsor of the document. Practice guidelines that serve to complement the current statement have been developed by the IDSA (3). In addition to the IDSA, representatives of the American Academy of Pediatrics (AAP), the (United States) National Tuberculosis Controllers Association (NTCA), the Canadian Thoracic Society (CTS), the IUATLD, and the WHO participated in the revision. By virtue of their different perspectives these committee members served to provide broader input and to help ensure that the guidelines are placed in an appropriate context. It should be emphasized that the current guidelines are intended for areas in which mycobacterial cultures, drug susceptibility tests, radiographic facilities, and second-line drugs are available, either immediately or by referral, on a routine basis. For this revision of the recommendations essentially all clinical trials of antituberculosis treatment in the English language literature were reviewed and the strength of the evidence they presented was rated according to the IDSA/USPHS rating scale (4). This revision of the recommendations for treatment of tuberculosis presents a significant philosophic departure from previous versions. In this document the responsibility for successful treatment of tuberculosis is placed primarily on the provider or program initiating therapy rather than on the patient. It is well established that appropriate treatment of tuberculosis rapidly renders the patient noninfectious, prevents drug resistance, minimizes the risk of disability or death from tuberculosis, and nearly eliminates the possibility of relapse. For these reasons, antituberculosis chemotherapy is both a personal and a public health measure that cannot be equated with the treatment of, for example, hypertension or diabetes mellitus, wherein the benefits largely accrue to the patient. Provider responsibility is a central concept in treating patients with tuberculosis, no matter what the source of their care. All reasonable attempts should be made to accommodate the patient so that a successful outcome is achieved. However, interventions such as detention may be necessary for patients who are persistently nonadherent. The recommendations in this statement are not applicable under all epidemiologic circumstances or across all levels of resources that are available to tuberculosis control programs worldwide. Although the basic principles of therapy described in this document apply regardless of conditions, the diagnostic approach, methods of patient supervision, and monitoring for response and for adverse drug effects, and in some instances the regimens recommended, are quite different in high-incidence, low-income areas compared with low-incidence, high-income areas of the world. A summary of the important differences between the recommendations in this document and those of the IUATLD and the WHO is found in Section 10,Treatment of Tuberculosis in Low-Income Countries: Recommendations of the WHO and the IUTLD. In the United States there has been a call for the elimination of tuberculosis, and a committee constituted by the Institute of Medicine (IOM) issued a set of recommendations for reaching this goal (5). The IOM committee had two main recommendations related to treatment of tuberculosis; first, that all U.S jurisdictions have health regulations that mandate completion of therapy (treatment until the patient is cured); and second, that all treatment be administered in the context of patient-centered programs that are based on individual patient characteristics and needs. The IOM recommendations emphasize the importance of the structure and organization of treatment services, as well as the drugs that are used, to treat patients effectively. This philosophy is the core of the DOTS strategy (described in Section 10 Treatment of Tuberculosis in Low-Income Countries: Recommendations oof the WHO and the IUTLD), developed by the IUATLD and implemented globally by the WHO. Thus, although there are superficial differences in the approach to tuberculosis treatment between high- and low-incidence countries, the fundamental concern, regardless of where treatment is given, is ensuring patient adherence to the drug regimen and successful completion of therapy (6). References

2. Organization and Supervision of TreatmentSuccessful treatment of tuberculosis depends on more than the science of chemotherapy. To have the highest likelihood of success, chemotherapy must be provided within a clinical and social framework based on an individual patient's circumstances. Optimal organization of treatment programs requires an effective network of primary and referral services and cooperation between clinicians and public health officials, between health care facilities and community outreach programs, and between the private and public sectors of medical care. This section describes the approaches to organization of treatment that serve to ensure that treatment has a high likelihood of being successful. As noted previously, antituberculosis chemotherapy is both a personal health measure intended to cure the sick patient and a basic public health strategy intended to reduce the transmission of Mycobacterium tuberculosis. Typically, tuberculosis treatment is provided by public health departments, often working in collaboration with other providers and organizations including private physicians, community health centers, migrant health centers, correctional facilities, hospitals, hospices, long-term care facilities, and homeless shelters. Private providers and public health departments may cosupervise patients, assuring that the patient completes therapy in a setting that is not only mutually agreeable but also enables access to tuberculosis expertise and resources that might otherwise not be available. In managed care settings delivery of tuberculosis treatment may require a more structured public/private partnership, often defined by a contract, to assure completion of therapy. Regardless of the means by which treatment is provided, the ultimate legal authority for assuring that patients complete therapy rests with the public health system. 2.1. Role of the Health Department The responsibility of the health department in the control of tuberculosis is to ensure that all persons who are suspected of having tuberculosis are identified and evaluated promptly and that an appropriate course of treatment is prescribed and completed successfully (1,2). A critical component of the evaluation scheme is access to proficient microbiological laboratory services, for which the health department is responsible. The responsibilities of the health department may be accomplished indirectly by epidemiologic surveillance and monitoring of treatment decisions and outcome, applying generally agreed-on standards and guidelines, or more directly by provision of diagnostic and treatment services, as well as by conducting epidemiologic investigations. Given the diverse sociodemographic characteristics of patients with tuberculosis and the many mechanisms by which health care is delivered, the means by which the goals of the health department are accomplished may be quite varied. In dealing with individual patients, approaches that focus on each person's needs and characteristics should be used to determine a tailored treatment plan that is designed to ensure completion of therapy (3). Such treatment plans are developed with the patient as an active participant together with the physician and/or nurse, outreach workers, social worker (when needed), and others as appropriate. Given that one-half the current incident cases of tuberculosis in the United States were born outside the United States (similar circumstances prevail in most other low-incidence countries), translation of materials into the patient's primary language is often necessary to ensure his/her participation in developing the treatment plan. Ideally, a specific case manager is assigned individual responsibility for assuring that the patient completes therapy. The treatment plan is reviewed periodically and revised as needed. These reviews may be accomplished in meetings between the patient and the assigned provider, as well as more formally through case and cohort evaluations. The treatment plan is based on the principle of using the least restrictive measures that are likely to achieve success. The full spectrum of measures that may be employed ranges from, at an absolute minimum, monthly monitoring of the patient in the outpatient setting to legally mandated hospitalization (4). Directly observed therapy (DOT) is the preferred initial means to assure adherence. For nonadherent patients more restrictive measures are implemented in a stepwise fashion. Any approach must be balanced, ensuring that the needs and rights of the patient, as well as those of the public, are met. Care plans for patients being managed in the private sector should be developed jointly by the health department and the private provider, and must address identified and anticipated barriers to adherence. 2.2. Promoting Adherence Louis Pasteur once said, "The microbe is nothing...the terrain everything" (5). Assuming appropriate drugs are prescribed, the terrain (the circumstances surrounding each patient that may affect his or her ability to complete treatment) becomes the most important consideration in completion of tuberculosis treatment. Many factors may be part of this terrain. Factors that interfere with adherence to the treatment regimen include cultural and linguistic barriers to cooperation, lifestyle differences, homelessness, substance abuse, and a large number of other conditions and circumstances that, for the patient, are priorities that compete with taking treatment for tuberculosis (6). Barriers may be patient related, such as conflicting health beliefs, alcohol or drug dependence, or mental illness, or they may be system related, such as lack of transportation, inconvenient clinic hours, and lack of interpreters (7). Effective tuberculosis case management identifies and characterizes the terrain and determines an appropriate care plan based on each of the identified factors. Additional advantages of the patient-centered approach are that, by increasing communication with the patient, it provides opportunities for further education concerning tuberculosis and enables elicitation of additional information concerning contacts. To maximize completion of therapy, patient-centered programs identify and utilize a broad range of approaches based on the needs and circumstances of individual patients. Among these approaches, DOT is the preferred initial strategy and deserves special emphasis. Although DOT itself has not been subjected to controlled trials in low-incidence areas (and, thus, is rated AII), observational studies and a meta-analysis in the United States strongly suggest that DOT, coupled with individualized case management, leads to the best treatment results (8--10). To date there have been three published studies of DOT in high-incidence areas, two of which (11,12) showed no benefit and one (13) in which there was a significant advantage for DOT. What is clear from these studies is that DOT cannot be limited merely to passive observation of medication ingestion; there must be aggressive interventions when patients miss doses. Using DOT in this manner can only improve results. DOT can be provided daily or intermittently in the office, clinic, or in the "field" (patient's home, place of employment, school, street corner, bar, or any other site that is mutually agreeable) by appropriately trained personnel. DOT should be used for all patients residing in institutional settings such as hospitals, nursing homes, or correctional facilities, or in other settings, such as methadone treatment sites, that are conducive to observation of therapy (14). However, even in such supervised settings careful attention must be paid to ensuring that ingestion of the medication is, in fact, observed. It is essential that all patients being treated with regimens that use intermittent drug administration have all doses administered under DOT because of the potentially serious consequences of missed doses. DOT also enables early identification of nonadherence, adverse drug reactions, and clinical worsening of tuberculosis. DOT provides a close connection to the health care system for a group of patients at high risk of other adverse health events and, thus, should facilitate identification and management of other conditions. The use of DOT does not guarantee ingestion of all doses of every medication (15). Patients may miss appointments, may not actually swallow the pills, or may deliberately regurgitate the medications. Consequently, all patients, including those who are being treated by DOT, should continue to be monitored for signs of treatment failure. DOT is only one aspect of a comprehensive patient-centered program that, in addition, includes incentives and enablers described subsequently (16--20). Patients who are more likely to present a transmission risk to others or are more likely to have problems with adherence (Table 7) should be prioritized for DOT when resources are limited. When DOT is not being used, fixed-dose combination preparations (see Section 6.2, Fixed-Dose Combination Preparations) containing INH and RIF or INH, RIF, and PZA reduce the risk of the patient taking only one drug and may help prevent the development of drug resistance. Combination formulations are easier to administer and also may reduce medication errors. Depending on the identified obstacles to completion of therapy, the treatment plan may also include enablers and incentives such as those listed in Table 8. Studies have examined the use of a patient-centered approach that utilizes DOT in addition to other adherence-promoting tools (9,21,22). These studies demonstrate, as shown in Figure 3, that "enhanced DOT" (DOT together with incentives and enablers) produces the highest treatment completion rates (in excess of 90% across a range of geographic and socioeconomic settings), and reinforces the importance of patient-related factors in designing and implementing case management (9,23). Intensive educational efforts should be initiated as soon as the patient is suspected of having tuberculosis. The instruction should be at an educational level appropriate for the patient and should include information about tuberculosis, expected outcomes of treatment, the benefits and possible adverse effects of the drug regimen, methods of supervision, assessment of response, and a discussion of infectiousness and infection control. The medication regimen must be explained in clear, understandable language and the verbal explanation followed with written instructions. An interpreter is necessary when the patient and health care provider do not speak the same language. Materials should be appropriate for the culture, language, age, and reading level of the patient. Relevant information should be reinforced at each visit. The patient's clinical progress and the treatment plan must be reviewed at least monthly to evaluate the response to therapy and to identify adherence problems. Use of a record system (Figure 4) either manual or computer-based, that quantifies the dosage and frequency of medication administered, indicates AFB smear and culture status, and notes symptom improvement as well as any adverse effects of treatment serves to facilitate the regular reviews and also provides data for cohort analyses. In addition, adherence monitoring by direct methods, such as the detection of drugs or drug metabolites in the patient's urine, or indirect methods, such as pill counts or a medication monitor, should be a part of routine management, especially if the patient is not being given DOT. Tracking patients is also a critical concern for those charged with assuring completion of treatment. It has been shown that patients who move from one jurisdiction to another before completion of therapy are much more likely to default than patients who do not move (24). Factors that have been shown to be associated with moving/defaulting include diagnosis of tuberculosis in a state correctional facility, drug and alcohol abuse, and homelessness. Communication and coordination of services among different sources of care and different health departments are especially important for patients in these groups as well as for migrant workers and other patients with no permanent home. Such communication may also be necessary across national boundaries, especially the United States--Mexico border, and there are systems in place to facilitate such communication and tracking. Some patients, for example those with tuberculosis caused by drug-resistant organisms, or who have comorbid conditions, such as HIV infection, alcoholism, or other significant underlying disorders, may need to be hospitalized in a facility where tuberculosis expertise is available and where there are appropriate infection control measures in place. Hospitalization may be necessary for nonadherent patients for whom less restrictive measures have failed (25--27). Public health laws exist in most states that allow the use of detainment under these circumstances, at least for patients who remain infectious (28). Court-ordered DOT has been used successfully in some states as a less costly alternative. The use of these interventions depends on the existence of appropriate laws, cooperative courts, and law enforcement officials, and the availability of appropriate facilities. Health departments must be consulted to initiate legal action when it is necessary. References
   3. Drugs in Current UseCurrently, there are 10 drugs approved by the United States Food and Drug Administration (FDA) for treating tuberculosis (Table 9). In addition, the fluoroquinolones, although not approved by the FDA for tuberculosis, are used relatively commonly to treat tuberculosis caused by drug-resistant organisms or for patients who are intolerant of some of the first-line drugs. Rifabutin, approved for use in preventing Mycobacterium avium complex disease in patients with HIV infection but not approved for tuberculosis, is useful for treating tuberculosis in patients concurrently taking drugs that have unacceptable interactions with other rifamycins. Amikacin and kanamycin, nearly identical aminoglycoside drugs used in treating patients with tuberculosis caused by drug-resistant organisms, are not approved by the FDA for tuberculosis. Of the approved drugs isoniazid (INH), rifampin (RIF), ethambutol (EMB), and pyrazinamide (PZA) are considered first-line antituberculosis agents and form the core of initial treatment regimens. Rifabutin and rifapentine may also be considered first-line agents under the specific situations described below. Streptomycin (SM) was formerly considered to be a first-line agent and, in some instances, is still used in initial treatment; however, an increasing prevalence of resistance to SM in many parts of the world has decreased its overall usefulness. The remaining drugs are reserved for special situations such as drug intolerance or resistance. The drug preparations available currently and the recommended doses are shown in Tables 3, 4, and 5. 3.1. First-Line Drugs 3.1.1. Isoniazid Role in treatment regimen. Isoniazid (INH) is a first-line agent for treatment of all forms of tuberculosis caused by organisms known or presumed to be susceptible to the drug. It has profound early bactericidal activity against rapidly dividing cells (1,2). Dose. See Table 3. Adults (maximum): 5 mg/kg (300 mg) daily; 15 mg/kg (900 mg) once, twice, or three times weekly. Children (maximum): 10--15 mg/kg (300 mg) daily; 20--30 mg/kg (900 mg) twice weekly (3). Preparations. Tablets (50 mg, 100 mg, 300 mg); syrup (50 mg/5 ml); aqueous solution (100 mg/ml) for intravenous or intramuscular injection. Adverse effects. Asymptomatic elevation of aminotransferases: Aminotransferase elevations up to five times the upper limit of normal occur in 10--20% of persons receiving INH alone for treatment of latent tuberculosis infection (4). The enzyme levels usually return to normal even with continued administration of the drug. Clinical hepatitis: (see Table 10.) Data indicate that the incidence of clinical hepatitis is lower than was previously thought. Hepatitis occurred in only 0.1--0.15% of 11,141 persons receiving INH alone as treatment for latent tuberculosis infection in an urban tuberculosis control program (5). Prior studies suggested a higher rate, and a meta-analysis of six studies estimated the rate of clinical hepatitis in patients given INH alone to be 0.6% (6--8). In the meta-analysis the rate of clinical hepatitis was 1.6% when INH was given with other agents, not including RIF. The risk was higher when the drug was combined with RIF, an average of 2.7% in 19 reports (8). For INH alone the risk increases with increasing age; it is uncommon in persons less than 20 years of age but is nearly 2% in persons aged 50--64 years (6). The risk also may be increased in persons with underlying liver disease, in those with a history of heavy alcohol consumption, and, data suggest, in the postpartum period, particularly among Hispanic women (9). Fatal hepatitis: A large survey estimated the rate of fatal hepatitis to be 0.023%, but more recent studies suggest the rate is substantially lower (10,11). The risk may be increased in women. Death has been associated with continued administration of INH despite onset of symptoms of hepatitis (12). Peripheral neurotoxicity (13,14): This adverse effect is dose related and is uncommon (less than 0.2%) at conventional doses (15--17). The risk is increased in persons with other conditions that may be associated with neuropathy such as nutritional deficiency, diabetes, HIV infection, renal failure, and alcoholism, as well as for pregnant and breastfeeding women. Pyridoxine supplementation (25 mg/day) is recommended for patients with these conditions to help prevent this neuropathy (18). Central nervous system effects: Effects such as dysarthria, irritability, seizures, dysphoria, and inability to concentrate have been reported but have not been quantified. Lupus-like syndrome (19): Approximately 20% of patients receiving INH develop anti-nuclear antibodies. Less than 1% develop clinical lupus erythematosis, necessitating drug discontinuation. Hypersensitivity reactions: Reactions, such as fever, rash, Stevens-Johnson syndrome, hemolytic anemia, vasculitis, and neutropenia are rare. Monoamine (histamine/tyramine) poisoning: This has been reported to occur after ingestion of foods and beverages with high monoamine content but is rare (20--22). If flushing occurs, patients should be instructed to avoid foods and drinks, such as certain cheeses and wine, having high concentrations of monoamines. Diarrhea: Use of the commercial liquid preparation of INH, because it contains sorbitol, is associated with diarrhea. Use in pregnancy. INH is considered safe in pregnancy, but the risk of hepatitis may be increased in the peripartum period (9,23). Pyridoxine supplementation (25 mg/day) is recommended if INH is administered during pregnancy (18). It should be noted that multivitamin preparations have variable amounts of pyridoxine but generally less than 25 mg/day and, thus, do not provide adequate supplementation. CNS penetration. Penetration is excellent. Cerebrospinal fluid (CSF) concentrations are similar to concentrations achieved in serum (24). Use in renal disease. (See Section 8.7: Renal Insufficiency and End-Stage Renal Disease.) INH can be used safely without dose adjustment in patients with renal insufficiency (25) and with end-stage renal isease who require chronic hemodialysis (26). Use in hepatic disease. (See Section 8.8: Hepatic Disease.) The risk of drug accumulation and drug-induced hepatitis may be increased in the presence of hepatic disease; however, INH may be used in patients with stable hepatic disease. Laboratory and clinical monitoring should be more frequent in such situations. Monitoring. Routine monitoring is not necessary. However, for patients who have preexisting liver disease or who develop abnormal liver function that does not require discontinuation of the drug, liver function tests should be measured monthly and when symptoms occur. Serum concentrations of phenytoin and carbamazepine may be increased in persons taking INH. However, in combination therapy with RIF the effects of INH on serum concentrations of the anticonvulsants are limited by the decrease caused by RIF. Thus, it is important to measure serum concentrations of these drugs in patients receiving INH with or without RIF and adjust the dose if necessary. 3.1.2. Rifampin Role in treatment regimen. Rifampin (RIF) is a first-line agent for treatment of all forms of tuberculosis caused by organisms with known or presumed sensitivity to the drug. It has activity against organisms that are dividing rapidly (early bactericidal activity) (1) and against semidormant bacterial populations, thus accounting for its sterilizing activity (27). Rifampin is an essential component of all short-course regimens. Dose. See Table 3. Adults (maximum): 10 mg/kg (600 mg) once daily, twice weekly, or three times weekly. Children (maximum): 10--20 mg/kg (600 mg) once daily or twice weekly. Preparations. Capsules (150 mg, 300 mg); contents of capsule may also be mixed in an appropriate diluent to prepare an oral suspension; aqueous solution for parenteral administration. Adverse effects (28). Cutaneous reactions (29): Pruritis with or without rash may occur in as many as 6% of patients but is generally self-limited (30). This reaction may not represent true hypersensitivity and continued treatment with the drug may be possible. More severe, true hypersensitivity reactions are uncommon, occurring in 0.07--0.3% of patients (17,31,32). Gastrointestinal reactions (nausea, anorexia, abdominal pain): The incidence is variable, but symptoms are rarely severe enough to necessitate discontinuation of the drug (28--30). Flulike syndrome: This may occur in 0.4--0.7% of patients receiving 600 mg twice weekly but not with daily administration of the same dose (31--34). Symptoms are more likely to occur with intermittent administration of a higher dose (29,35). Hepatotoxicity: Transient asymptomatic hyperbilirubinemia may occur in as many as 0.6% of patients receiving the drug. More severe clinical hepatitis that, typically, has a cholestatic pattern may also occur (8,36). Hepatitis is more common when the drug is given in combination with INH (2.7%) than when given alone (nearly 0%) or in combination with drugs other than INH (1.1%) (8). Severe immunologic reactions: In addition to cutaneous reactions and flulike syndrome, other reactions thought to be immune mediated include the following: thrombocytopenia, hemolytic anemia, acute renal failure, and thrombotic thrombocytopenic purpura. These reactions are rare, each occurring in less than 0.1% of patients (31,32,37). Orange discoloration of bodily fluids (sputum, urine, sweat, tears): This is a universal effect of the drug. Patients should be warned of this effect at the time treatment is begun. Soft contact lenses and clothing may be permanently stained. Drug interactions due to induction of hepatic microsomal enzymes: There are a number of drug interactions (described in Section 7, Drug Interactions, and Table 12) with potentially serious consequences. Of particular concern are reductions, often to ineffective levels, in serum concentrations of common drugs, such as oral contraceptives, methadone, and warfarin. In addition there are important bidirectional interactions between rifamycins and antiretroviral agents. Because information regarding rifamycin drug interactions is evolving rapidly, readers are advised to consult the CDC web site www.cdc.gov/nchstp/tb/ to obtain the most up-to-date information. Use in pregnancy. RIF is considered safe in pregnancy (38). CNS penetration. Concentrations in the CSF may be only 10--20% of serum levels, but this is sufficient for clinical efficacy. Penetration may be improved in the setting of meningitis (39). Use in renal disease. (See Section 8.7: Renal Insufficiency and End-Stage Renal Disease.) RIF can be used safely without dose adjustment in patients with renal insufficiency and end-stage renal disease (26,40). Use in hepatic disease. (see Section 8.8: Hepatic Disease.) Clearance of the drug may be impaired in the presence of liver disease, causing increased serum levels (40). However, because of the critical importance of rifampin in all short-course regimens, it generally should be included, but the frequency of clinical and laboratory monitoring should be increased. Monitoring. No routine monitoring tests are required. However, rifampin causes many drug interactions described in Section 7, Drug Interactions, that may necessitate regular measurements of the serum concentrations of the drugs in question. 3.1.3. Rifabutin Role in treatment regimen. Rifabutin is used as a substitute for RIF in the treatment of all forms of tuberculosis caused by organisms that are known or presumed to be susceptible to this agent. The drug is generally reserved for patients who are receiving any medication having unacceptable interactions with rifampin (41) or have experienced intolerance to rifampin. Dose. See Table 3. Adults (maximum): 5 mg/kg (300 mg) daily, twice, or three times weekly. The dose may need to be adjusted when there is concomitant use of protease inhibitors or nonnucleoside reverse transcriptase inhibitors. When rifabutin is used with efavirenz the dose of rifabutin should be increased to 450--600 mg either daily or intermittently. Because information regarding rifamycin drug interactions is evolving rapidly readers are advised to consult the CDC web site, http://www.cdc.gov/nchstp/tb/, to obtain the most up-to-date information. Children (maximum): Appropriate dosing for children is unknown. Preparations: Capsules (150 mg) for oral administration. Adverse effects. Hematologic toxicity: In a placebo-controlled, double-blind trial involving patients with advanced acquired immunodeficiency syndrome (AIDS) (CD4+ cell counts <200 cells/µl), neutropenia occurred in 25% compared with 20% in patients receiving placebo (p = 0.03). Neutropenia severe enough to necessitate discontinuation of the drug occurred in 2% of patients receiving the drug (product insert B; Adria Laboratories, Columbus, OH). The effect is dose related, occurring more frequently with daily than with intermittent administration of the same dose (42). In several studies of patients with and without HIV infection, neither neutropenia nor thrombocytopenia was associated with rifabutin (43--47). Uveitis: This is a rare (less than 0.01%) complication when the drug is given alone at a standard (300 mg daily) dose. The occurrence is higher (8%) with higher doses or when rifabutin is used in combination with macrolide antimicrobial agents that reduce its clearance (48). Uveitis may also occur with other drugs that reduce clearance such as protease inhibitors and azole antifungal agents. Gastrointestinal symptoms: These symptoms occurred in 3% of patients with advanced HIV infection given 300 mg/day (package insert). In subsequent studies no increased incidence of gastrointestinal symptoms was noted among patients taking rifabutin (43,44,46--48). Polyarthralgias: This symptom occurred in 1--2% of persons receiving a standard 300-mg dose (package insert). It is more common at higher doses (48). Polyarthralgias have not been noted in more recent studies involving both HIV-infected and uninfected patients (43,44,46,47). Hepatotoxity: Asymptomatic elevation of liver enzymes has been reported at a frequency similar to that of RIF (48). Clinical hepatitis occurs in less than 1% of patients receiving the drug. Pseudojaundice (skin discoloration with normal bilirubin): This is usually self-limited and resolves with discontinuation of the drug (49). Rash: Although initially reported to occur in as many as 4% of patients with advanced HIV infection, subsequent studies suggest that rash is only rarely (less than 0.1%) associated with rifabutin (46). Flulike syndrome: Flulike syndrome is rare (less than 0.1%) in patients taking rifabutin. Orange discoloration of bodily fluids (sputum, urine, sweat, tears): This is a universal effect of the drug. Patients should be warned of this effect at the time treatment is begun. Soft contact lenses and clothing may be permanently stained. Use in pregnancy. There are insufficient data to recommend the use of rifabutin in pregnant women; thus, the drug should be used with caution in pregnancy. CNS penetration. The drug penetrates inflamed meninges (50). Use in renal disease. (See Section 8.7: Renal Insufficiency and End-Stage Renal Disease.) Rifabutin may be used without dosage adjustment in patients with renal insufficiency and end-stage renal disease (50). Use in hepatic disease. (See Section 8.8: Hepatic Disease.) The drug should be used with increased clinical and laboratory monitoring in patients with underlying liver disease. Dose reduction may be necessary in patients with severe liver dysfunction (50). Monitoring. Monitoring is similar to that recommended for rifampin. Although drug interactions are less problematic with rifabutin, they still occur and close monitoring is required. 3.1.4. Rifapentine Role in treatment regimen. Rifapentine may be used once weekly with INH in the continuation phase of treatment for HIV-seronegative patients with noncavitary, drug-susceptible pulmonary tuberculosis who have negative sputum smears at completion of the initial phase of treatment (51). Dose. See Table 3. Adults (maximum): 10 mg/kg (600 mg), once weekly during the continuation phase of treatment. Data have suggested that a dose of 900 mg is well tolerated but the clinical efficacy of this dose has not been established (52). Children: The drug is not approved for use in children. Preparation. Tablet (150 mg, film coated). Adverse effects. The adverse effects of rifapentine are similar to those associated with RIF. Rifapentine is an inducer of multiple hepatic enzymes and therefore may increase metabolism of coadministered drugs that are metabolized by these enzymes (see Section 7: Drug Interactions). Use in pregnancy. There is not sufficient information to recommend the use of rifapentine for pregnant women. CNS penetration. There are no data on CSF concentrations of rifapentine. Use in renal disease. (See Section 8.7: Renal Insufficiency and End-Stage Renal Disease .) The pharmacokinetics of rifapentine have not been evaluated in patients with renal impairment. Although only about 17% of an administered dose is excreted via the kidneys, the clinical significance of impaired renal function in the disposition of rifapentine is not known. Use in hepatic disease. (See Section 8.8: Hepatic Disease.) The pharmacokinetics of rifapentine and its 25-desacetyl metabolite were similar among patients with various degrees of hepatic impairment and not different from those in healthy volunteers, even though the elimination of these compounds is primarily via the liver (53). The clinical significance of impaired hepatic function in the disposition of rifapentine and its 25-desacetyl metabolite is not known. Monitoring. Monitoring is similar to that for RIF. Drug interactions involving rifapentine are being investigated and are likely to be similar to those of RIF. 3.1.5. Pyrazinamide Role in treatment regimen. Pyrazinamide (PZA) is a first-line agent for the treatment of all forms of tuberculosis caused by organisms with known or presumed susceptibility to the drug. The drug is believed to exert greatest activity against the population of dormant or semidormant organisms contained within macrophages or the acidic environment of caseous foci (54). Adults: 20--25 mg/kg per day. Recommended adult dosages by weight, using whole tablets, are listed in Table 4. Children (maximum): 15--30 mg/kg (2.0 g) daily; 50 mg/kg twice weekly (2.0 g). Preparations. Tablets (500 mg, scored). Adverse effects. Hepatotoxicity: Early studies (55,56) using doses of 40--70 mg/kg per day reported high rates of hepatotoxicity. However, in treatment trials with multiple other drugs, including INH, liver toxicity has been rare at doses of 25 mg/kg per day or less (15,34,57). In one study, however, hepatotoxicity attributable to PZA used in standard doses occurred at a rate of about 1% (58). Gastrointestinal symptoms (nausea, vomiting): Mild anorexia and nausea are common at standard doses. Vomiting and severe nausea are rare except at high doses (59). Nongouty polyarthralgia: Polyarthralgias may occur in up to 40% of patients receiving daily doses of PZA. This rarely requires dosage adjustment or discontinuation of the drug (60). The pain usually responds to aspirin or other nonsteroidal antiinflammatory agents. In clinical trials of PZA in the initial intensive phase of treatment, athralgias were not noted to be a significant problem (15,61). Asymptomatic hyperuricemia: This is an expected effect of the drug and is generally without adverse consequence (15,62). Acute gouty arthritis: Acute gout is rare except in patients with preexisting gout (63), generally a contraindication to the use of the drug. Transient morbilliform rash: This is usually self-limited and is not an indication for discontinuation of the drug. Dermatitis: PZA may cause photosensitive dermatitis (59). Use in pregnancy. There is little information about the safety of PZA in pregnancy. However, when there are sound reasons to utilize a 6-month course of treatment, the benefits of PZA may outweigh the possible (but unquantified) risk. The WHO and the IUATLD recommend this drug for use in pregnant women with tuberculosis (see Section 10: Treatment of Tuberculosis in Low-Income Countries: Recommendations of the WHO and the IUATLD). CNS penetration. The drug passes freely into the CSF, achieving concentrations equivalent to those in serum (64). Use in renal disease. (See Section 8.7: Renal Insufficiency and End-Stage Renal Disease.) PZA is cleared primarily by the liver, but its metabolites are excreted in the urine and may accumulate in patients with renal insufficiency (65). The dose may, therefore, need to be reduced in patients with renal insufficiency. It should be administered at a reduced dose (25--35 mg/kg) three times a week after dialysis in patients with end-stage renal disease (Table 15) (26). The risk of hyperuricemia caused by PZA is increased in patients with renal insufficiency. Use in hepatic disease. (See Section 8.8: Hepatic Disease.) Although the frequency is slightly lower than with INH or RIF, the drug can cause liver injury that may be severe and prolonged. If the drug is used in patients with underlying liver disease, laboratory and clinical monitoring should be increased. Monitoring. Serum uric acid measurements are not recommended as a routine but may serve as a surrogate marker for compliance. Liver chemistry monitoring should be performed when the drug is used in patients with underlying liver disease or when it is used with rifampin in treating latent tuberculosis infection. 3.1.6. Ethambutol Role in treatment regimen. Ethambutol (EMB) is a first-line drug for treating all forms of tuberculosis. It is included in initial treatment regimens primarily to prevent emergence of RIF resistance when primary resistance to INH may be present. Ethambutol is generally not recommended for routine use in children whose visual acuity cannot be monitored. However, if a child has adult-type tuberculosis or disease that is suspected or proven to be caused by organisms that are resistant to either INH or RIF, EMB should be used (see Section 8.2: Children and Adolescents, and Table 6). Adults: 15--20 mg/kg per day: Table 5 lists recommended dosages for adults, using whole tablets. Children (maximum): 15--20 mg/kg per day (2.5 g); 50 mg/kg twice weekly (2.5 g). The drug can be used safely in older children but should be used with caution in children in whom visual acuity cannot be monitored (generally less than 5 years of age) (66). In younger children EMB can be used if there is concern with resistance to INH or RIF (Table 6). Preparations. Tablets (100 mg, 400 mg) for oral administration. Adverse effects. Retrobulbar neuritis: This is manifested as decreased visual acuity or decreased red-green color discrimination that may affect one or both eyes. The effect is dose related, with minimal risk at a daily dose of 15 mg/kg (67). No difference was found in the prevalence of decreased visual acuity between regimens that contained EMB at 15 mg/kg and those not containing the drug (68). The risk of optic toxicity is higher at higher doses given daily (18% of patients receiving more than 30 mg/kg per day) and in patients with renal insufficiency. Higher doses can be given safely twice or three times weekly. Peripheral neuritis: This is a rare adverse effect (69). Cutaneous reactions: Skin reactions requiring discontinuation of the drug occur in 0.2--0.7% of patients (68). Use in pregnancy. EMB is considered safe for use in pregnancy (70--72). CNS penetration. The agent penetrates the meninges in the presence of inflammation but does not have demonstrated efficacy in tuberculous meningitis (73). Use in renal disease. (See Section 8.7: Renal Insufficiency and End-Stage Renal Disease.) EMB is cleared primarily by the kidneys. The dose or dosing interval should be adjusted when the creatinine clearance is less than 70 ml/minute (74). EMB should be administered at a dose of 15--20 mg/kg three times a week by DOT after dialysis in patients with end-stage renal disease (Table 15) (26). Use in hepatic disease. (See Section 8.8: Hepatic Disease.) EMB can be used safely in patients with hepatic disease. Monitoring. Patients should have baseline visual acuity testing (Snellen chart) and testing of color discrimination (Ishihara tests). At each monthly visit patients should be questioned regarding possible visual disturbances including blurred vision or scotomata. Monthly testing of visual acuity and color discrimination is recommended for patients taking doses greater than 15--25 mg/kg, patients receiving the drug for longer than 2 months, and any patient with renal insufficiency. Patients should be instructed to contact their physician or public health clinic immediately if they experience a change in vision. EMB should be discontinued immediately and permanently if there are any signs of visual toxicity. 3.1.7. Fixed-dose combination preparations Role in treatment regimen. Two combined preparations, INH and RIF (Rifamate®) and INH, RIF, and PZA (Rifater®), are available in the United States. These formulations are a means of minimizing inadvertent monotherapy, particularly when DOT is not possible, and, therefore, may decrease the risk of acquired drug resistance (75). The use of fixed-dose formulations may reduce the number of pills that must be taken daily. Constituent drugs are combined in proportions compatible with daily treatment regimens. Formulations for intermittent administration are not available in the United States. Preparations and dose. Rifamate®: As sold in North America, each capsule contains RIF (300 mg) and INH (150 mg); thus, the daily dose is two capsules (600 mg of RIF and 300 mg of INH). Two capsules of Rifamate® plus two 300-mg tablets of INH are used by some programs for intermittent therapy given twice weekly as DOT. Rifater®: Each tablet contains RIF (120 mg), INH (50 mg), and PZA (300 mg). The daily dose is based on weight as follows: 44 kg or less, four tablets; 45--54 kg, five tablets; 55 kg or more, six tablets. To obtain an adequate dose of PZA in persons weighing more than 90 kg additional PZA tablets must be given. Adverse effects. See comments under individual drugs above. Use in pregnancy. Rifamate® may be used in daily treatment of pregnant women. Rifater® should not be used because it contains PZA. CNS penetration. See comments under individual drugs above. Use in renal disease. (See Section 8.7: Renal Insufficiency and End-Stage Renal Disease.) Rifamate® may be used in persons with renal insufficiency. Rifater® should not be used because of the potential need for adjustment of the dose of PZA. Use in hepatic disease. (See Section 8.8: Hepatic Disease.) In patients with underlying hepatic disease it is advisable to treat with single-drug formulations until safety in an individual patient can be determined and a stable regimen established. 3.2. Second-Line Drugs 3.2.1. Cycloserine Role in treatment regimen. Cycloserine (76,77) is a second-line drug that is used for treating patients with drug-resistant tuberculosis caused by organisms with known or presumed susceptibility to the agent. It may also be used on a temporary basis for patients with acute hepatitis in combination with other nonhepatotoxic drugs. Dose. See Table 3. Adults (maximum): 10--15 mg/kg per day (1,000 mg), usually 500--750 mg/day given in two doses. Clinicians with experience with cycloserine indicate that toxicity is more common at doses over 500 mg/day. Serum concentration measurements aiming for a peak concentration of 20--35 mg/ml are often useful in determining the optimum dose for a given patient. There are no data to support intermittent administration. Children (maximum): 10--15 mg/kg per day (1.0 g/day). Preparations. Capsules (250 mg). Adverse effects. Central nervous system effects: The central nervous system effects range from mild reactions, such as headache or restlessness, to severe reactions, such as psychosis and seizures. The drug may exacerbate underlying seizure disorders or mental illness. Seizures have been reported to occur in up to 16% of patients receiving 500 mg twice daily but in only 3% when receiving 500 mg once daily (78). Pyridoxine may help prevent and treat neurotoxic side effects and is usually given in a dosage of 100--200 mg/day (79). Rarely, cycloserine may cause peripheral neuritis. Use in pregnancy. Cycloserine crosses the placenta. There are limited data on safety in pregnancy; thus, it should be used in pregnant women only when there are no suitable alternatives (77). CNS penetration. Concentrations in CSF approach those in serum (77). Use in renal disease. (See Section 8.7: Renal Insufficiency and End-Stage Renal Disease.) The drug can accumulate in patients with impaired renal function and should be used cautiously in such patients. Generally, the dose should be reduced and serum concentrations measured. Cycloserine should not be used in patients having a creatinine clearance of less than 50 ml/minute unless the patient is receiving hemodialysis. For patients being hemodialyzed the dose should be 500 mg three times a week or 250 mg daily (Table 15). Serum concentrations of the drug should be measured and the dose adjusted accordingly. Use in hepatic disease. (See Section 8.8: Hepatic Disease.) There are no precautions except for patients with alcohol-related hepatitis in whom there is an increased risk of seizures (77). Monitoring. Neuropsychiatric status should be assessed at least at monthly intervals and more frequently if symptoms develop. As noted above, measurements of serum concentrations may be necessary until an appropriate dose is established. For patients taking phenytoin, serum concentrations of phenytoin should be measured. 3.2.2. Ethionamide Role in treatment. Ethionamide (76,77) is a second-line drug that is used for patients with drug-resistant tuberculosis disease caused by organisms that have demonstrated or presumed susceptibility to the drug. Dose: See Table 3. Adults (maximum): 15--20 mg/kg per day (1.0 g/day), usually 500--750 mg/day in a single daily dose or two divided doses. The single daily dose can be given at bedtime or with the main meal. There are no data to support intermittent dosing. Children (maximum): 15--20 mg/kg per day (1.0 g/day). Preparations: Tablets (250 mg). Adverse reactions. Gastrointestinal effects: Ethionamide commonly causes profound gastrointestinal side effects, including a metallic taste, nausea, vomiting (that is often severe), loss of appetite, and abdominal pain (80). Symptoms may improve if doses are taken with food or at bedtime. Hepatotoxicity: Ethionamide is similar in structure to INH and may cause similar side effects. Hepatotoxicity occurs in about 2% of patients taking the drug (81,82). Neurotoxicity: Neurotoxicity, including peripheral neuritis, optic neuritis, anxiety, depression, and psychosis, has been reported in 1--2% of patients taking shorter courses of the drug with higher rates reported with prolonged treatment (83,84). Endocrine effects: Endocrine disturbances, including gynecomastia, alopecia, hypothyroidism, and impotence, have been described (85,86). Diabetes may be more difficult to manage in patients taking ethionamide (77). Use in pregnancy. Ethionamide crosses the placenta and is teratogenic in laboratory animals. It should not be used in pregnancy. CNS penetration. CSF concentrations are equal to those in serum (77). Use in renal disease. (See Section 8.7: Renal Insufficiency and End-stage Renal Disease.) For patients having a creatinine clearance of less than 30 ml/minute or who are receiving hemodialysis the dose should be reduced to 250--500 mg/day (Table 15). Use in hepatic disease. (See Section 8.8: Hepatic Disease.) Ethionamide should be used with caution in patients with underlying liver disease. Monitoring. Liver function tests should be obtained at baseline and, if there is underlying liver disease, at monthly intervals. The studies should be repeated if symptoms occur. Thyroid-stimulating hormone should be measured at baseline and at monthly intervals. 3.2.3. Streptomycin Role in treatment regimen. Streptomycin (SM) (76,77,87--89) and EMB have been shown to be approximately equivalent when used in the initial phase of treatment with 6-month regimens. However, among patients likely to have acquired M. tuberculosis in a high-incidence country, the relatively high rate of resistance to SM limits its usefulness. Dose. See Table 3. Adults (maximum): 15 mg/kg per day (1 g/day) parenterally, usually given as a single daily dose (5--7 days/week) initially, and then reducing to two or three times a week after the first 2--4 months or after culture conversion, depending on the efficacy of the other drugs in the regimen (90). For persons over 59 years of age, the dose should be reduced to 10 mg/kg per day (750 mg). The dosing frequency should be reduced (i.e., 12--15 mg/kg per dose two or three times per week) in persons with renal insufficiency (see below: Use in Renal Disease) (91,92). Children (maximum): 20--40 mg/kg per day (1 g/day). Preparations. Aqueous solution in vials of 1 g (93). Adverse effects. Ototoxicity: The most important adverse reaction caused by SM is ototoxicity, including vestibular and hearing disturbances. The risk is increased with age (94) or concomitant use of loop-inhibiting diuretics (furosemide, ethacrynic acid). The risk of ototoxicity increases with increasing single doses and with the cumulative dose, especially above 100--120 g. Neurotoxicity: SM relatively commonly causes circumoral parasthesias immediately after injection. Rarely, it may interact with muscle relaxants to cause postoperative respiratory muscle weakness. Nephrotoxicity: Nephrotoxicity occurs less commonly with SM than with amikacin, kanamycin, or capreomycin (95). Renal insufficiency requiring discontinuation occurs in about 2% of patients (96). Use in pregnancy. SM is contraindicated in pregnancy because of the risk of fetal hearing loss (77,97,98). CNS penetration. There is only slight diffusion of SM into CSF, even in patients with meningitis (77,99) Use in renal disease. (See Section 8.7: Renal Insufficiency and End-Stage Renal Disease.) SM should be used with caution in patients with renal function impairment because of the increased risk of both ototoxicity and nephrotoxicity. Because clearance is almost exclusively by the kidney, dosing adjustments are essential in patients with underlying renal insufficiency, including the elderly and those undergoing hemodialysis. In such patients, the dosing frequency should be reduced to two or three times weekly, but the milligram dose should be maintained at 12--15 mg/kg per dose to take advantage of the concentration-dependent bactericidal effect (Table 15) (91,92). Smaller doses may reduce the efficacy of this drug. The drug should be given after dialysis to facilitate DOT and to avoid premature removal of the drug (100). Serum drug concentrations should be monitored to avoid toxicity (91). Use in hepatic disease. (See Section 8.8: Hepatic Disease.) No precautions are necessary. Monitoring. An audiogram, vestibular testing, Romberg testing, and serum creatinine measurement should be performed at baseline. Assessments of renal function, and questioning regarding auditory or vestibular symptoms, should be performed monthly. An audiogram and vestibular testing should be repeated if there are symptoms of eighth nerve toxicity. 3.2.4. Amikacin and kanamycin Role in treatment regimen. Amikacin and kanamycin (76,77,101) are two closely related injectable second-line drugs that are used for patients with drug-resistant tuberculosis whose isolate has demonstrated or presumed susceptibility to the agents. There is nearly always complete cross-resistance between the two drugs, but most SM-resistant strains are susceptible to both (102). Because it is used to treat a number of other types of infections, amikacin may be more easily obtained, and serum drug concentration measurements are readily available. Dose. See Table 3. Adults (maximum): 15 mg/kg per day (1.0 g/day), intramuscular or intravenous, usually given as a single daily dose (5--7 days/week) initially, and then reducing to two or three times a week after the first 2--4 months or after culture conversion, depending on the efficacy of the other drugs in the regimen (90). For persons greater than 59 years of age the dose should be reduced to 10 mg/kg per day (750 mg). The dosing frequency should be reduced (i.e., 12--15 mg/kg per dose, two or three times per week) in persons with renal insufficiency (see below: Use in Renal Disease) (91,92). Children (maximum): 15--30 mg/kg per day (1 g/day) intramuscular or intravenous as a single daily dose. Preparations. Aqueous solution for intramuscular or intravenous injection in vials of 500 mg and 1 g. Adverse effects. Ototoxicity: Amikacin and kanamycin may cause deafness, but they cause less vestibular dysfunction than SM (103,104). Ototoxicity is more common with concurrent use of diuretics. In one report high-frequency hearing loss occurred in 24% of patients receiving amikacin, with higher rates occurring among those receiving longer treatment and/or higher doses (105), whereas a review of the literature found only 1.5% hearing loss (106). Nephrotoxicity: Amikacin and kanamycin may be more nephrotoxic than SM (95). Renal impairment was seen in 8.7% of patients receiving amikacin, with a higher frequency in patients with initially increased creatinine levels, patients receiving larger total doses, and patients receiving other nephrotoxic agents. A frequency of 3.4% was reported in patients with no risk factors (106,107). Use in pregnancy. Both amikacin and kanamycin are contraindicated in pregnant women because of risk of fetal nephrotoxicity and congenital hearing loss (77). CNS penetration. Only low concentrations of the drugs are found in CSF, although slightly higher concentrations have been found in the presence of meningitis (77). Use in renal disease. (See Section 8.7: Renal Insufficiency and End-Stage Renal Disease.) Amikacin and kanamycin should be used with caution in patients with renal function impairment because of the increased risk of both ototoxicity and nephrotoxicity. Because clearance is almost exclusively by the kidney, dosing adjustments are essential in patients with underlying renal insufficiency, including the elderly and those receiving hemodialysis. In such patients, the dosing frequency should be reduced to two or three times per week, but the dose should be maintained at 12--15 mg/kg to take advantage of the concentration-dependent bactericidal effect (Table 15) (91,92). Smaller doses may reduce the efficacy of this drug. The drug should be given after dialysis to facilitate DOT and to avoid premature removal of the drug (100). Serum drug concentrations should be monitored to avoid toxicity (91). Use in hepatic disease. (See Section 8.8: Hepatic Disease.) No precautions are necessary. Monitoring. Monitoring should be performed as described for SM. An advantage of amikacin is that serum concentration measurements can be obtained routinely. Patients with severe hepatic disease, because of predisposition to hepato-renal syndrome, may be at greater risk for nephrotoxicity from amikacin/kanamycin and should have renal function monitored closely. 3.2.5. Capreomycin Role in treatment. Capreomycin is a second-line injectable drug that is used for patients with drug-resistant tuberculosis caused by organisms that have known or presumed susceptibility to the drug (108). Dose. See Table 3. Adults (maximum): 15 mg/kg per day (1.0 g/day), usually given as a single daily dose five to seven times a week, and reduced to two or three times a week after the first 2--4 months or after culture conversion, depending on the efficacy of the other drugs in the regimen (90). For persons greater than 59 years of age the dose should be reduced to 10 mg/kg per day (750 mg). The dosing frequency should be reduced to 12--15 mg/kg two or three times per week in persons with renal insufficiency (see below: Use In Renal Disease) (91,92). Children (maximum): 15--30 mg/kg per day (1 g/day) as a single daily or twice weekly dose. Preparations. Capreomycin is available in vials of 1 g for both intramuscular and intravenous administration. Adverse effects. Nephrotoxicity: Nephrotoxic effects may result in reduced creatinine clearance or potassium and magnesium depletion. Proteinuria is common (109). Significant renal toxicity requiring discontinuation of the drug has been reported to occur in 20--25% of patients (110,111). Ototoxicity: Vestibular disturbances, tinnitus, and deafness appear to occur more often in elderly persons or those with preexisting renal impairment (111). Use in pregnancy. Capreomycin should be avoided in pregnancy because of risk of fetal nephrotoxicity and congenital hearing loss (77). CNS penetration. Capreomycin does not penetrate into the CSF (77). Use in renal disease. (see Section 8.7: Renal Insufficiency and End-Stage Renal Disease.) Capreomycin should be used with caution in patients with renal function impairment because of the increased risk of both ototoxicity and nephrotoxicity (112). Because capreomycin is nearly entirely cleared by the kidneys, dosing adjustments are essential in patients with underlying renal insufficiency and end-stage renal disease, including patients undergoing hemodialysis. In such patients, the dosing frequency should be reduced to two or three times weekly, but the milligram dose should be maintained at 12--15 mg/kg per dose to take advantage of the concentration-dependent bactericidal effect (Table 15) (91,92). Smaller doses may reduce the efficacy of this drug. The drug should be given after dialysis to facilitate DOT and avoid premature removal of the drug (100,113). Serum drug concentrations should be monitored to avoid toxicity (91). Use in hepatic disease. (See Section 8.8: Hepatic Disease.) No precautions are necessary. Monitoring. Monitoring should be performed as described for SM. In addition, serum potassium and magnesium concentrations should be measured at baseline and at least at monthly intervals. 3.2.6. p-Aminosalicylic acid Role in treatment. p-Aminosalicylic acid (PAS) is an oral agent used in treatment of drug-resistant tuberculosis caused by organisms that are susceptible to the drug. Dose. See Table 3. Adults: 8--12 g/day in two or three doses. For PAS granules, 4 g three times daily has been the usual dosage (114,115). However, it has been shown that administration of 4 g twice daily is adequate to achieve the target serum concentration (116). Children: 200--300 mg/kg per day in two to four divided doses (117). Preparations. The only available formulation in the United States is granules in 4-g packets (Paser Granules®) (118). It was previously thought that the granules needed to be taken with acidic food (115); however, more recent data suggest that this is not necessary (C. Peloquin, personal communication). Tablets (500 mg) are still available in some countries. A solution for intravenous administration is available in Europe (119,120). Adverse effects. Hepatotoxicity: In a review of 7,492 patients being treated for tuberculosis, 38 (0.5%) developed hepatitis, of which 28 cases (0.3%) were attributed at least in part to PAS (121). Gastrointestinal distress: This is the most common side effect of PAS (122). In a large study of INH and PAS 11% of patients had drug toxicity, mainly gastrointestinal intolerance to PAS (114). The incidence of gastrointestinal side effects is less with lower doses (8 g daily) and with the granular formulation of the drug. Malabsorption syndrome: This is characterized by steatorrhea and low serum folate levels (123). Hypothyroidism: This is a common side effect, especially with prolonged administration or concomitant use of ethionamide. It may be accompanied by goiter formation. Thyroid hormone replacement may be required. Thyroid function returns to normal after discontinuation of the drug (124). Coagulopathy: A doubling of the prothrombin time that seemed to be lessened by coadministration of streptomycin has been reported (125). Use in pregnancy. No studies have been done in humans; however, PAS has been used safely in pregnancy. The drug should be used only if there are no alternatives (see below) for a pregnant woman who has multidrug-resistant tuberculosis. CNS penetration. In the presence of inflamed meninges, PAS concentrations are between 10--50% of those achieved in serum (119). The drug has marginal efficacy in meningitis. Use in renal disease. (See Section 8.7: Renal Insufficiency and End-Stage Renal Disease.) Approximately 80% of the drug is excreted in the urine (118). Unless there is no alternative, PAS is contraindicated in severe renal insufficiency because of the accumulation of the acetylated form (123,126,127). Because both PAS and acetyl-PAS are removed by dialysis, the drug should be given after dialysis to facilitate DOT and avoid premature removal of the drug (126). Use in hepatic disease. (See Section 8.8: Hepatic Disease.) The clearance of PAS is not substantially altered in liver disease, suggesting that the drug may be used in usual doses but with increased laboratory and clinical monitoring (127). Monitoring. Hepatic enzymes and thyroid function should be measured at baseline. With prolonged therapy (i.e., more than 3 months) thyroid function should be checked every 3 months. 3.2.7. Fluoroquinolones Role in treatment regimen. Of the fluoroquinolones (128--131), levofloxacin, moxifloxacin, and gatifloxacin have the most activity against M. tuberculosis. On the basis of cumulative experience suggesting a good safety profile with long-term use of levofloxacin, this drug is the preferred oral agent for treating drug-resistant tuberculosis caused by organisms known or presumed to be sensitive to this class of drugs, or when first-line agents cannot be used because of intolerance. Data on long-term safety and tolerability of moxifloxacin and gatifloxacin, especially at doses above 400 mg/day, are limited. Cross-resistance has been demonstrated among ciprofloxacin, ofloxacin, and levofloxacin and presumably is a class effect (132). Fluoroquinolones should not be considered first-line agents for the treatment of drug-susceptible tuberculosis except in patients who are intolerant of first-line drugs. Dose. (See Table 3.) The doses given are for levofloxacin. Adults: 500--1,000 mg daily. Children: The long-term (more than several weeks) use of fluoroquinolones in children and adolescents has not been approved because of concerns about effects on bone and cartilage growth. However, most experts agree that the drug should be considered for children with MDR tuberculosis. The optimal dose is not known. Preparations (Levofloxacin). Tablets (250 mg, 500 mg, 750 mg); aqueous solution (500 mg) for intravenous administration. Adverse effects. The adverse effects (133) cited are for levofloxacin. Gastrointestinal disturbance: Nausea and bloating occur in 0.5--1.8% of patients taking the drug. Neurologic effects: Dizziness, insomnia, tremulousness, and headache occur in 0.5% of patients. Cutaneous reactions: Rash, pruritis, and photosensitivity occur in 0.2--0.4% of patients. Use in pregnancy. This class of drugs should be avoided in pregnancy because of teratogenic effects (119,134). CNS penetration. The concentration in CSF after administration of a standard dose of levofloxacin is 16--20% of that in serum (135). Interference with absorption. Because antacids and other medications containing divalent cations markedly decrease absorption of fluoroquinolones, it is critical that any fluoroquinolone not be administered within 2 hours of such medications (see Section 7.1: Interactions Affecting Antituberculosis Drugs). Use in renal disease. (See Section 8.7: Renal Insufficiency and End Stage Renal Disease.) The drug is cleared primarily (80%) by the kidney (135). Dosage adjustment (750--1,000 mg three times a week) is recommended if creatinine clearance is less than 50 ml/minute (Table 15) (136). It is not cleared by hemodialysis; supplemental doses after dialysis are not necessary (135). Use in hepatic disease. Drug levels are not affected by hepatic disease (135). It is presumed to be safe for use in the setting of severe liver disease, but as with all drugs, should be used with caution.
References
  4. Principles of Antituberculosis Chemotherapy4.1. Combination Chemotherapy The primary goals of antituberculosis chemotherapy are to kill tubercle bacilli rapidly, prevent the emergence of drug resistance, and eliminate persistent bacilli from the host's tissues to prevent relapse (1). To accomplish these goals, multiple antituberculosis drugs must be taken for a sufficiently long time. The theoretical model of chemotherapy for tuberculosis is founded on current understanding of the biology of M. tuberculosis in the host and on the specific activities of antituberculosis drugs. This model is supported by data from numerous in vivo and in vitro studies. It is theorized that there are three separate subpopulations of M. tuberculosis within the host. These populations are defined by their growth characteristics and the milieu in which they are located (1). The largest of the subpopulations consists of rapidly growing extracellular bacilli that reside mainly in cavities. This subpopulation, because of its size, is most likely to harbor organisms with random mutations that confer drug resistance. The frequency of these mutations that confer resistance is about 10-6 for INH and SM, 10-8 for RIF, and 10-5 for EMB; thus, the frequency of concurrent mutations to both INH and RIF, for example, would be 10-14, making simultaneous resistance to both drugs in an untreated patient a highly unlikely event (2). INH has been shown to possess the most potent ability to kill rapidly multiplying M. tuberculosis during the initial part of therapy (early bactericidal activity), thereby rapidly decreasing infectiousness (3--5). It is followed in this regard by EMB, RIF, and SM. PZA has weak early bactericidal activity during the first 2 weeks of treatment (3,6). Drugs that have potent early bactericidal activity reduce the chance of resistance developing within the bacillary population. Early experience in clinical trials demonstrated that multiple agents are necessary to prevent the emergence of a drug-resistant population as a consequence of the selection pressure from administration of a single agent. Shortly after the discovery of SM, it was demonstrated that treatment with this agent alone resulted in treatment failure and drug resistance (7). Subsequently, it was shown that the combination of PAS and SM substantially lessened the likelihood of acquired resistance and treatment failure (8). In modern regimens both INH and RIF have considerable ability to prevent the emergence of drug resistance when given with another drug. EMB and SM are also effective in preventing the emergence of drug resistance, whereas the activity of PZA in this regard is poor (9,10). For this reason PZA should not be used with only one other agent when treating active tuberculosis. The rapidly dividing population of bacilli is eliminated early in effective therapy as shown by the early clinical responses and clearing of live bacilli from sputum within 2 months in about 80% of patients. The remaining subpopulations of M. tuberculosis account for treatment failures and relapses, especially when the duration of therapy is inadequate. These residual populations include organisms that are growing more slowly, often in the acidic environment provided by areas of necrosis, and a group that is characterized by having spurts of growth interspersed with periods of dormancy. The sterilizing activity of a drug is defined by its ability to kill bacilli, mainly in these two subpopulations that persist beyond the early months of therapy, thus decreasing the risk of relapse (1). The use of drugs that have good sterilizing properties is essential for regimens as short as 6 months. RIF and PZA have the greatest sterilizing activity followed by INH and SM (11,12). The sterilizing activity of RIF persists throughout the course of therapy, but this does not appear to be true for PZA. When given in RIF-containing regimens, PZA provides additive sterilizing activity only during the initial 2 months of therapy. The sterilizing activity of PZA may not be so limited in regimens where RIF cannot be used or is not effective, so regimens for MDR tuberculosis may include PZA for the full course of treatment if the isolate is susceptible to this agent. 4.2. Optimum Duration of Treatment Truly effective chemotherapy for tuberculosis became available with the introduction of INH in the early 1950s. Adding INH to SM and PAS increased cure rates from about 70 to 95% but required treatment for 18--24 months (13). Eventually, EMB replaced PAS as the companion agent for INH (14). Subsequent investigations of combination chemotherapy sought to identify regimens that were shorter and that could be given intermittently. The British Medical Research Council (BMRC) in East Africa (15) conducted the first large-scale multicenter study of short-course (6-month) regimens. This study demonstrated that the addition of RIF or PZA to a base regimen of daily SM and INH increased the proportion of patients whose sputum cultures were negative by 2 months after the initiation of treatment and significantly reduced the relapse rate. Moreover, the relapse rate of the short-course regimens was no greater than that of the standard 18-month regimen containing SM, INH, and thiacetazone (a drug used in many countries in place of PAS or EMB). In Hong Kong, administration of a 9-month regimen of SM, INH, and PZA daily, twice weekly, or three times weekly was associated with a relapse rate of only 5--6% (16). Unfortunately, all short-course regimens that did not include RIF required fully supervised therapy and SM had to be used for the entire 9 months. Subsequent investigations conducted by the British Thoracic Association demonstrated that SM (or EMB) was necessary only for the first 2 months to achieve excellent results with a 9-month treatment duration, using INH and RIF throughout (17,18). The BMRC conducted studies in Hong Kong proving that EMB was roughly as effective as SM in the initial phase of therapy, thereby demonstrating that an all-oral regimen was effective (19). The addition of PZA to a regimen containing INH and RIF enabled further shortening of the duration of therapy to 6 months. The British Thoracic Association demonstrated that a regimen of INH and RIF for 6 months, supplemented during the first 2 months with PZA and either EMB or SM, was as effective as a 9-month regimen of INH and RIF with EMB in the first 2 months (18). Administration of PZA beyond the initial 2 months in an RIF-containing regimen had no additional benefit. The efficacy of the treatment regimens was similar regardless of whether PZA was given for 2, 4, or 6 months (20). Subsequent studies of 6-month regimens have served to refine the approach used currently. USPHS Trial 21 compared self-administered INH and RIF for 6 months plus PZA given during the initial 2 months with INH and RIF for 9 months (21). EMB was added only if INH resistance was suspected. Patients taking the 6-month PZA-containing regimen had negative sputum cultures sooner after treatment was started than those treated for 9 months without PZA and relapse rates were similar for the two regimens (3.5 versus 2.8%). Investigators in Denver reported a low relapse rate (1.6%) when using a 62-dose, directly observed, 6-month regimen that consisted of 2 weeks of daily INH, RIF, PZA, and SM, 6 weeks of the same four drugs given twice weekly, and 18 weeks of twice weekly INH and RIF (22). Regimens less than 6 months in duration have been shown to have unacceptably high relapse rates among patients with smear-positive pulmonary tuberculosis (23,24). However, in a study in Hong Kong among patients with smear-negative, culture-positive tuberculosis, the relapse rate was about 2% when using a 4-month regimen of daily SM, INH, RIF, and PZA (25); among smear-negative, culture-negative cases, the relapse rate was only 1%. In Arkansas, patients with tuberculosis who had negative smears and cultures were treated with INH and RIF given daily for 1 month followed by 3 months of twice weekly INH and RIF (26). Only 3 of 126 (2.4%) patients developed active tuberculosis during 3.5 years of follow-up. Thus, it appears that a 4-month, INH- and RIF-containing regimen is effective in culture-negative tuberculosis (see Section 8.4: Culture-Negative Pulmonary Tuberculosis in Adults). 4.3. Intermittent Drug Administration Nonadherence to the antituberculosis treatment regimen is well known to be the most common cause of treatment failure, relapse, and the emergence of drug resistance. Administration of therapy on an intermittent basis, as opposed to daily dosing, facilitates supervision of therapy, thereby improving the outcome. The concept of intermittent administration of antituberculosis drugs developed from early clinical observations and was supported by subsequent laboratory investigations. First, it was noted that a single daily dose of 400 mg of INH was more effective than the same total dose given in two divided doses (27). Second, in an early study from Madras, investigators demonstrated that fully supervised twice weekly therapy could be delivered to nonhospitalized patients and that the results were better than with a conventional self-administered daily regimen (28). These findings, plus the laboratory results noted below, led to a series of clinical trials that compared daily and intermittent dosing of antituberculosis medications. In all of these studies, intermittent regimens were demonstrated to be as effective as daily regimens and no more toxic (20). In the laboratory it was noted that in vitro exposure of tubercle bacilli to drugs was followed by a lag period of several days before growth began again (postantibiotic effect) (29--31). Thus, it was concluded that maintaining continuous inhibitory drug concentrations was not necessary to kill or inhibit growth of M. tuberculosis. Studies in guinea pigs substantiated that INH could be given at intervals as long as 4 days without loss of efficacy; however, there was a significant decrease in activity with an 8-day dosing interval (30,31). The concept of intermittent drug administration continues to evolve. Studies have demonstrated that the frequency of drug administration in the continuation phase of treatment may be decreased to once a week when using INH and rifapentine for certain highly selected patients (32--34). Because of the newness of these findings the data are presented in some detail. The results from three open-label, randomized clinical trials indicate that rifapentine given with INH once a week is safe and effective when used for the treatment of selected, HIV-negative patients with pulmonary tuberculosis. In a study performed in Hong Kong, patients with pulmonary tuberculosis were allocated at random to receive 600 mg of rifapentine and 900 mg of INH given either once every week or once every 2 of 3 weeks for 4 months after completion of a standard 2-month initial phase (32). Overall, about 11% of patients in the two rifapentine arms failed or relapsed during a 5-year follow-up period, compared with 4% of the patients who received three times weekly INH--RIF (control arm) in the continuation phase of treatment. Omitting every third dose of INH--rifapentine did not appreciably increase the relapse rate, indicating that modest nonadherence may have a negligible effect. Multivariate analyses showed that the significant prognostic factors were treatment arm, radiographic extent of disease (all three regimens), and sex (women fared better than men). The frequency of failures and relapses was also greater in all three arms if the 2-month culture was positive. The pivotal study for drug registration was conducted in North America and South Africa among HIV-negative patients with pulmonary tuberculosis (33). Patients in the experimental arm received directly observed twice weekly rifapentine together with daily self-administered INH, PZA, and EMB in the initial 2 months, followed by 4 months of once weekly directly observed rifapentine and INH. Patients in the control arm received a standard four-drug initial phase, followed by twice weekly INH--RIF. Relapse rates during 2 years of follow-up were similar to those seen in the Hong Kong study (8.2% relapse in the experimental arm versus 4.4% in the control arm), and cavitary disease, sputum culture positivity at the end of the initial phase, and nonadherence with INH, EMB, and PZA in the experimental arm were significantly associated with an increased probability of relapse. The third study was conducted by the CDC Tuberculosis Trials Consortium, and employed a design similar to the Hong Kong trial, in which HIV-negative patients were allocated at random after successful completion of standard 2-month initial phase therapy (34). Again, results, as measured by rates of failure/relapse, were remarkably similar to the first two trials, 9.2% in the experimental (INH--rifapentine once weekly) arm compared with 5.6% in the control (INH--RIF twice weekly) arm. However, as in the South Africa study, relapse was significantly associated with the presence of cavitary lesions seen on the initial chest film and sputum culture positivity at 2 months, both of which were more common in the rifapentine arm. With adjustment for these factors, the difference in outcome in the two arms was not statistically significant. Relapse rates among patients who did not have cavitary disease and had negative sputum cultures at 2 months were low in both treatment arms. However, in patients who had both cavitation and a positive culture at 2 months the relapse rate in the rifapentine arm was 22% and in the twice weekly INH--RIF arm was 21% (Table 11). In all of the cited studies, rifapentine was well tolerated, with the adverse events being similar to those occurring with RIF. A small number of HIV--positive patients were enrolled in the CDC study, but this arm was closed after the development of acquired rifampin resistance among relapse cases in the rifapentine arm (35). References
 5. Recommended Treatment Regimens5.1. Evidence-based Rating System To assist in making informed treatment decisions based on the most credible research results, evidence-based ratings have been assigned to the treatment recommendations (Table 1). The ratings system is the same as that used in the recommendations for treating latent tuberculosis infection, in which a letter indicating the strength of the recommendation, and a roman numeral indicating the quality of the evidence supporting the recommendation, are assigned to each regimen (1). Thus, clinicians can use the ratings to differentiate among recommendations based on data from clinical trials and those based on the opinions of experts familiar with the relevant clinical practice and scientific rationale for such practice when clinical trial data are not available. 5.2. Recommended Regimens There are four basic regimens recommended for treating adults with tuberculosis caused by organisms that are known or presumed to be susceptible to INH, RIF, PZA, and EMB (Table 2). As noted below, children, depending on the circumstances, may not receive EMB in the initial phase of a 6-month regimen, but the regimens are otherwise identical. Each regimen has an initial phase of 2 months, followed by a choice of several options for the continuation phase of either 4 or 7 months. In Table 2 the initial phase is denoted by a number (1, 2, 3, or 4) and the options for the continuation phase are denoted by the respective number and a letter designation (a, b, or c). DOT is the preferred initial management strategy for all regimens and should be used whenever feasible. All patients being given drugs less than 7 days per week (5, 3, or 2 days/week) must receive DOT. 5.2.1. Six-month regimens The current minimal acceptable duration of treatment for all children and adults with culture-positive tuberculosis is 6 months (26 weeks). The initial phase of a 6-month regimen for adults should consist of a 2-month period of INH, RIF, PZA, and EMB given daily throughout (Regimen 1), daily for 2 weeks followed by two times weekly for 6 weeks (Regimen 2), or three times a week (Regimen 3). The minimum number of doses is specified in Table 2. On the basis of substantial clinical experience, 5 day-a-week drug administration by DOT is considered to be equivalent to 7 day-a-week administration; thus, either may be considered "daily." Although administration of antituberculosis drugs by DOT at 5 days/week, rather than 7 days, has been reported in a large number of studies it has not been compared with 7-day administration in a clinical trial and therefore is rated AIII. The recommendation that a four-drug regimen be used initially for all patients is based on the current proportion of new tuberculosis cases caused by organisms that are resistant to INH (2). This recommendation is supported by a retrospective analysis of data from various BMRC studies indicating that in the presence of INH resistance there were fewer treatment failures and relapses if a regimen containing four drugs, INH, RIF, PZA, and EMB, was used in the initial phase (3). However, if therapy is being initiated after drug susceptibility test results are known and the organisms are susceptible to INH and RIF, EMB is not necessary. EMB can be discontinued as soon as the results of drug susceptibility studies demonstrate that the isolate is susceptible to the first-line agents. In most situations these results are not available before 6--8 weeks after treatment is begun. The continuation phase of treatment should consist of INH and RIF given for a minimum of 4 months (18 weeks). Patients should be treated until they have received the specified total number of doses for the treatment regimen (Table 2). The continuation phase can be given daily (Regimen 1a), twice weekly (Regimens 1b and 2a), or three times weekly (Regimen 3a). The continuation phase should be extended for an additional 3 months for patients who have cavitation on the initial or follow-up chest radiograph and are culture-positive at the time of completion of the initial phase of treatment (2 months). Patients who are HIV negative, who do not have cavities on the chest radiograph, and who have negative sputum AFB smears at completion of the initial phase of treatment may be treated with once weekly INH and rifapentine in the continuation phase for 4 months. If the culture of the sputum obtained at 2 months is positive, observational data and expert opinion suggest that the continuation phase of once weekly INH and rifapentine should be 7 months (4). 5.2.2. Nine-month regimen If PZA cannot be included in the initial regimen, or if the isolate is determined to be resistant to PZA (an unusual circumstance, except for Mycobacterium bovis and M. bovis var. BCG), a regimen consisting of INH, RIF, and EMB should be given for the initial 2 months (Regimen 4) followed by INH and RIF for 7 months given either daily or twice weekly (Regimens 4a and 4b). 5.2.3. Alternative regimens In some cases, either because of intolerance or drug resistance, the above-described regimens cannot be used. In these instances, an alternative regimen may be required. In a retrospective analysis of the combined results of clinical trials conducted by the BMRC it was concluded that, in the presence of initial resistance to INH, if a four-drug regimen containing RIF and PZA was used in the initial phase and RIF was used throughout a 4-month continuation phase there were no treatment failures and 7% relapses compared with 4% relapses among patients with fully susceptible strains (3). Data from a Hong Kong BMRC study suggest that in the presence of INH resistance results are better when PZA is used throughout (5). On the basis of these data, when INH cannot be used or the organisms are resistant to INH, a 6-month regimen of RIF, PZA, and EMB is nearly as efficacious as an INH-containing regimen (Rating BI) (3). Alternatively, RIF and EMB for 12 months may be used, preferably with PZA during at least the initial 2 months (Rating BII) (5,6). If RIF is not used, INH, EMB, and FQN should be given for a minimum of 12--18 months supplemented with PZA during at least the initial 2 months (Rating BIII). An injectable agent may also be included for the initial 2--3 months for patients with more extensive disease or to shorten the duration (e.g., to 12 months), (7,8). Levofloxacin, moxifloxacin, or gatifloxacin may be useful in alternative regimens, but the potential role of a fluoroquinolone and optimal length of therapy have not been defined (9,10). In situations in which several of the first-line agents cannot be used because of intolerance, regimens based on the principles described for treating multiple drug-resistant tuberculosis (Section 9.3: Management of Tuberculosis Caused by Drug-Resistant Organisms) should be used. 5.3. Deciding to Initiate Treatment The decision to initiate combination chemotherapy for tuberculosis should be based on epidemiologic information, clinical and radiographic features of the patient, and the results of the initial series of AFB smears (preferably three) and, subsequently, cultures for mycobacteria. Rapid amplification tests, if used, can also confirm the diagnosis of tuberculosis more quickly than cultures. On the basis of this information, the likelihood that a given patient has tuberculosis can be estimated. For example, a patient who has emigrated recently from a high-incidence country, has a history of cough and weight loss, and has characteristic findings on chest radiograph should be considered highly likely to have tuberculosis. In such situations combination drug therapy should be initiated, even before AFB smear and mycobacterial culture results are known. Empirical treatment with a four-drug regimen should be initiated promptly when a patient is seriously ill with a disorder that is thought possibly to be tuberculosis. Initiation of treatment should not be delayed because of negative AFB smears for patients in whom tuberculosis is suspected and who have a life-threatening condition. Disseminated (miliary) tuberculosis, for example, is often associated with negative sputum AFB smears. Likewise, for a patient with suspected tuberculosis and a high risk of transmitting M. tuberculosis if, in fact, she or he had the disease, combination chemotherapy should be initiated in advance of microbiological confirmation of the diagnosis to minimize potential transmission. A positive AFB smear provides strong inferential evidence for the diagnosis of tuberculosis. If the diagnosis is confirmed by isolation of M. tuberculosis or a positive nucleic acid amplification test, or is strongly inferred from clinical or radiographic improvement consistent with a response to treatment, the regimen can be continued to complete a standard course of therapy (Figure 1). A PPD-tuberculin skin test may be done at the time of initial evaluation, but a negative test does not exclude the diagnosis of active tuberculosis. However, a positive skin test supports the diagnosis of culture-negative pulmonary tuberculosis or, in persons with stable abnormal chest radiographs consistent with inactive tuberculosis, a diagnosis of latent tuberculosis infection (see below). If the cultures are negative, the PPD-tuberculin skin test is positive (5 mm or greater induration), and there is no response to treatment, the options are as follows: 1) stop treatment if RIF and PZA have been given for at least 2 months; 2) continue treatment with RIF, with or without INH, for a total of 4 months; or 3) continue treatment with INH for a total of 9 months (11). All three of these options provide adequate therapy for persons with prior tuberculosis once active disease has been excluded. If clinical suspicion for active tuberculosis is low, the options are to begin treatment with combination chemotherapy or to defer treatment until additional data have been obtained to clarify the situation (usually within 2 months) (Figure 2, top). Even when the suspicion of active tuberculosis is low, treatment for latent tuberculosis infection with a single drug should not be initiated until active tuberculosis has been excluded. In low-suspicion patients not initially treated, if cultures remain negative, the PPD-tuberculin skin test is positive (5 mm or greater induration), and the chest radiograph is unchanged after 2 months, there are three treatment options (Figure 2, top) (11). The preferred options are INH for 9 months or RIF, with or without INH, for 4 months. RIF and PZA for a total of 2 months can be used for patients not likely to complete a longer regimen and who can be monitored closely. However, this last regimen has been associated with an increased risk of hepatotoxicity and should be used only in the limited circumstances described (12,13). An advantage of the early use of combination chemotherapy is that, once active disease is excluded by negative cultures and lack of clinical or radiographic response to treatment, the patient will have completed 2 months of combination treatment that can be applied to the total duration of treatment recommended for latent tuberculosis infection (Figure 2, bottom). 5.4. Baseline and Follow-Up Evaluations Patients suspected of having tuberculosis should have appropriate specimens collected for microscopic examination and mycobacterial culture. When the lung is the site of disease, three sputum specimens should be obtained 8--24 hours apart. In patients who are not producing sputum spontaneously, induction of sputum using aerosolized hypertonic saline or bronchoscopy (performed under appropriate infection control procedures) may be necessary to obtain specimens. Susceptibility testing for INH, RIF, and EMB should be performed on an initial positive culture, regardless of the source. Second-line drug susceptibility testing should be done only in reference laboratories and be limited to specimens from patients who have had prior therapy, have been in contact of a patient with known drug resistance, have demonstrated resistance to rifampin or two other first-line drugs, or who have positive cultures after more than 3 months of treatment. At the time treatment is initiated, in addition to the microbiologic examinations, it is recommended that all patients with tuberculosis have counseling and testing for HIV infection (14). Patients with epidemiologic factors suggesting a risk for hepatitis B or C, for example, injection drug use, birth in Asia or Africa, or HIV infection, should have serologic tests for these viruses (15,16). HIV-infected patients should also undergo CD4+ lymphocyte count measurement. Measurements of AST, bilirubin, alkaline phosphatase, and serum creatinine and a platelet count should be obtained for all adults. Testing of visual acuity (Snellen chart) and color vision (Ishihara tests) should be performed when EMB is to be used. During treatment of patients with pulmonary tuberculosis, at a minimum, a sputum specimen for AFB smear and culture should be obtained at monthly intervals until two consecutive specimens are negative on culture. As described subsequently, important decisions concerning the continuation-phase regimen hinge on the microbiological status at the end of the initial phase of treatment, thus, obtaining sputum specimens at this juncture is critical, if sputum conversion to negative has not already been documented. For patients who had positive AFB smears at the time of diagnosis, follow-up smears may be obtained at more frequent intervals (e.g., every 2 weeks until two consecutive specimens are negative) to provide an early assessment of the response to treatment, especially for patients in situations in which the risk of transmission is high. On occasion, AFB-positive sputa are culture negative; this occurs most frequently among patients with far advanced cavitary tuberculosis after the first months of treatment. It is thought that these organisms are dead and that their presence is not a sign of treatment failure, even if noted later in treatment. However, repeat cultures should be obtained to confirm that the earlier culture result was correct and not a false negative. Drug susceptibility tests should be repeated on isolates from patients who have positive cultures after 3 months of treatment. As described in Section 9.2 (Treatment Failure), patients who have positive cultures after 4 months of treatment should be considered as having failed treatment and managed accordingly. For patients with extrapulmonary tuberculosis the frequency and kinds of evaluations will depend on the sites involved and the ease with which specimens can be obtained. In addition to the microbiological evaluations, it is essential that patients have clinical evaluations at least monthly to identify possible adverse effects of the antituberculosis medications and to assess adherence. For patients with positive cultures at diagnosis, a repeat chest radiograph at completion of 2 months of treatment may be useful but is not essential. A chest radiograph at completion of therapy provides a baseline against which subsequent examinations can be compared, but, as with the 2-month examination, it is not essential. When the initial sputum cultures are negative, a presumptive diagnosis can be made if radiographic improvement is noted, generally by the time 2 months of treatment has been completed. Thus, in patients with negative initial cultures, a chest radiograph is necessary after 2 months of treatment and a radiograph at completion of treatment is desirable. Generally, follow-up after completion of therapy is not necessary. As a routine, it is not necessary to monitor liver or renal function or platelet count for patients being treated with first-line drugs unless there were abnormalities at baseline or there are clinical reasons to obtain the measurements. Patients who have stable abnormalities of hepatic or renal function at baseline should have repeat measurements early in the course of treatment, then less frequently to ensure that there has not been worsening. Patients receiving EMB should be questioned regarding visual disturbances at monthly intervals; monthly repeat testing of visual acuity and color vision is recommended for patients receiving an EMB dose exceeding 15--20 mg/kg (the recommended range) and for patients receiving the drug for more than 2 months. Monitoring tests for the individual second-line drugs are listed in Section 3: Drugs in Current Use. 5.5. Identification and Management of Patients at Increased Risk of Relapse The result of a sputum culture at the conclusion of the initial phase of treatment (2 months) has been shown to correlate with the likelihood of relapse after completion of treatment for pulmonary tuberculosis. In seven clinical trials performed by the BMRC, the regimens that had the highest proportion of patients with a positive sputum culture at 2 months after treatment was initiated were associated with a higher likelihood of relapse within 2 years (17). Of greater relevance to the current recommendations, data from USPHS Trial 22 comparing once weekly rifapentine and INH with twice weekly RIF and INH, showed an increased rate of relapse in patients who had a positive culture at 2 months in both study arms (18). Cavitation on the initial chest radiograph was also an independent risk factor for relapse. In patients in the control arm (twice weekly INH--RIF) the presence of both cavitation and a positive culture at completion of 2 months of therapy was associated with a 21% rate of relapse, compared with 2% for patients who had neither risk factor (Table 11). Similar findings were reported in a retrospective analysis of data from BMRC trials (17) and from a USPHS trial conducted in Poland (19). The most effective means of decreasing the likelihood of relapse for patients at increased risk has not yet been determined by clinical trials. However, in a controlled trial of treatment for silicotuberculosis in Hong Kong, prolongation of the continuation phase from 4 to 6 months decreased the rate of relapse from 22 to 7% (p <0.025) (20). Also in studies from Hong Kong, it was found that increasing the duration of PZA beyond the 2-month initial phase did not improve the efficacy of RIF-containing regimens (21). It has been reported that for patients at high risk of relapse, prolongation of the once weekly INH--rifapentine continuation phase from 4 to 7 months resulted in significantly better results compared with patients in an earlier trial (4). In view of this evidence and on the basis of expert opinion, it is recommended that treatment for patients who have cavitation noted on the initial chest radiograph and who have positive cultures at completion of 2 months of therapy should be extended with INH and RIF for an additional 3 months for a total of 9 months (Rating AIII). In USPHS Study 22 patients treated with INH and RIF twice weekly in the continuation phase who had either cavitation on the initial chest radiograph or a positive culture at 2 months had approximately a 5--6% rate of relapse (Table 11) (18). This rate of adverse outcomes is not deemed to be sufficient to recommend prolongation of the continuation phase; however, patients with one or the other of these risk factors should be monitored more closely and consideration given to lengthening treatment if there are suggestions of a poor response. Additional factors to be considered in deciding to prolong treatment in patients with either cavitation or a positive culture at 2 months (but not both) might include being more than 10% underweight at diagnosis, having HIV infection, or having extensive involvement on chest radiograph. Patients with noncavitary pulmonary tuberculosis and a negative AFB smear at 2 months who are started on the once weekly rifapentine--INH continuation phase and are subsequently found to be culture positive at 2 months should have treatment extended by an additional 3 months for a total of 9 months. 5.6. Definition of Completion of Therapy Treatment for a defined duration without accounting for the number of doses taken can result in undertreatment. Therefore, the determination of whether or not treatment has been completed is based on the total number of doses taken---not solely on the duration of therapy (Table 2). For example, the 6-month daily (given 7 days/week) regimen should consist of at least 182 doses of INH and RIF, and 56 doses of PZA. If the drugs are administered by DOT at 5 days/week, the minimum number of doses is 130. A similar reduction in the target number of doses for 5-day-a-week administration applies to any of the regimens with a daily component. In some cases, either because of drug toxicity or nonadherence to the regimen, the specified number of doses cannot be administered within the targeted time period. In such cases, it is recommended that all of the specified number of doses for the initial phase be delivered within 3 months and those for the 4-month continuation phase be delivered within 6 months, so that the 6-month regimen should be completed within 9 months. If these targets are not met the patient must be considered to have interrupted therapy and be managed as described below. 5.7. Interruptions in Therapy Interruptions in therapy are common in the treatment of tuberculosis. When interruptions occur, the person responsible for supervision must decide whether to restart a complete course of treatment or simply to continue as intended originally. This decision depends in part on whether the interruption occurred during the initial or the continuation phase of therapy. In general, the earlier the break in therapy and the longer its duration, the more serious the effect and the greater the need to restart the treatment from the beginning. Continuous treatment is more important in the initial phase of therapy, when there is the highest bacillary population and the chance of developing drug resistance is greatest. During the continuation phase, the number of bacilli is much smaller and the goal of therapy is to kill the persisting organisms. The duration of the interruption and the bacteriological status of the patient before and after the interruption are also important considerations. There is no evidence on which to base detailed recommendations for managing interruptions in treatment, and no recommendations will cover all of the situations that may arise. The following approach (summarized in Figure 5), modified from the New York City Bureau of Tuberculosis Control Clinical Policies and Protocols (22), is presented as an example. If the interruption occurs during the initial phase of treatment and the lapse is 14 days or more in duration, treatment should be restarted from the beginning. However, if the lapse is less than 14 days, the treatment regimen should be continued. In either instance the total number of doses targeted for the initial phase should be given. If the interruption in treatment occurs during the continuation phase after the patient has received more than 80% of the planned total continuation phase doses given by DOT, further treatment may not be necessary if the patient's sputum was AFB smear negative on initial presentation. However, for patients who were smear positive initially, continued treatment to complete the planned total number of doses is warranted. If the patient has received less than 80% of the planned total doses and the lapse is 3 months or more in duration, treatment should be restarted from the beginning. If the lapse is less than 3 months in duration, treatment should be continued to complete a full course. At the time the patient is returned to treatment sputum cultures should be obtained and repeat drug susceptibility testing performed. If the cultures are still positive, the treatment regimen should be restarted. If sputum cultures are negative the patient could be treated as having culture-negative tuberculosis and given an additional 4 months of combination chemotherapy. Regardless of the timing and duration of the interruption, DOT should be used. If the patient was already being managed with DOT, additional measures will be necessary to ensure completion of therapy. Consultation with an expert is recommended to assist in managing treatment interruptions. References
 6. Practical Aspects of Treatment6.1. Drug Administration The first-line antituberculosis medications should be administered together as single dose rather than in divided doses. A single dose leads to higher, and potentially more effective, peak serum concentrations. Administering a single daily dose also facilitates using DOT. Ingestion with food delays or moderately decreases the absorption of antituberculosis drugs (1). However, given the wide therapeutic margin of the first-line agents, the effects of food are of little clinical significance. Thus, if patients have epigastric distress or nausea with the first-line drugs, dosing with food is recommended. Administration with food is preferable to splitting a dose or changing to a second-line drug. The absorption of INH can be substantially decreased when the drug is ingested with glucose or lactose. Because of this effect, the commercial preparation of INH elixir uses sorbitol for flavor, rather than glucose or lactose. However, sorbitol can cause diarrhea, limiting the acceptability of the commercial INH elixir. Administration of crushed INH tablets in a food with relatively low concentrations of glucose, such as applesauce, has not been formally evaluated, but has been used successfully by many providers. Antacids have minimal effects on the absorption of the first-line antituberculosis drugs. With the exception of fluoroquinolones, there is little information regarding the effect of food and antacids on the second-line antituberculosis drugs. In the absence of data, it is preferable to administer the drugs on an empty stomach if they are tolerated. However, antacids and other medications containing divalent cations markedly decrease the absorption of the fluoroquinolones, an interaction that has been associated with failure of antibiotic therapy (2,3). Therefore, it is critical that any fluoroquinolone not be administered within 2 hours of a dose of antacids, the chewable tablet form of didanosine, sucralfate, iron, magnesium, calcium, zinc, or vitamins or dietary supplements (e.g., Ensure®, Sustical®) containing a significant amount of these cations. Parenteral therapy is indicated for severely ill patients who cannot take oral therapy and may be useful for the uncommon patient for whom poor absorption has been documented. Preparations of INH, RIF, the aminoglycosides, capreomycin, and most fluoroquinolones are available for intravenous administration. 6.2. Fixed-Dose Combination Preparations There are two fixed-dose combination preparations currently available for use in the United States, a combination of INH and RIF (Rifamate®) and a combination of INH, RIF, and PZA (Rifater®) (see Section 3: Drugs in Current Use). (A four-drug combination of INH, RIF, EMB, and PZA is available in some countries.) Two tablets of Rifamate® provide conventional daily doses of both INH (300 mg) and RIF (600 mg). The Rifater® tablet that is available in the United States contains INH (50 mg), RIF (120 mg), and PZA (300 mg). Six tablets of Rifater® would provide INH (300 mg) RIF (720 mg), and PZA (1,800 mg). The RIF dose is higher than is used typically in the United States because the RIF is less bioavailable in this formulation. These fixed-dose combinations have been formulated for use in daily therapy, although some programs use Rifamate® plus INH tablets for twice weekly treatment. It should be noted that the dose of PZA in Rifater® is such that additional PZA tablets will be required to provide an adequate dose for persons weighing more than 90 kg. Although there is no evidence indicating that fixed-dose combination medications are superior to individual drugs, expert opinion suggests that these formulations should be used when DOT is given daily and when DOT is not possible. Moreover, they are strongly recommended in international recommendations of the WHO and IUATLD. The theoretical advantage of reducing the risk of inadvertent monotherapy, the ease of administration, and the potential for reducing medication errors make them preferable to individual medications in many instances. When prescribing a fixed-dose combination preparation, care must be taken because of the similarity of the trade names of RIF (Rifadin®) and the fixed-dose combinations (Rifamate®, Rifater®). 6.3. Management of Common Adverse Effects As is true with all medications, combination chemotherapy for tuberculosis is associated with a predictable incidence of adverse effects, some mild, some serious. A comprehensive list of reported adverse reactions and their frequency is described in Section 3: Drugs in Current Use. Mild adverse effects can generally be managed with symptomatic therapy, whereas with more severe effects the offending drug or drugs must be discontinued. Although it is important to be attuned to the potential for adverse effects it is at least equally important that first-line drugs not be stopped without adequate justification. The following is a summary, based largely on clinical experience and expert opinion, of the approaches that should be taken in managing the common adverse effects of tuberculosis treatment. Proper management of more serious adverse reactions often requires expert consultation. 6.3.1. Gastrointestinal upset: nausea, vomiting, poor appetite, abdominal pain Gastrointestinal reactions are common, particularly in the first few weeks of therapy. Many of the antituberculosis drugs can cause gastrointestinal upset (4). In the presence of gastrointestinal symptoms serum AST and bilirubin should be measured. If the AST level is less than three times the upper limit of normal, the symptoms are assumed not to be due to hepatic toxicity. However, if the AST level is three or more times the upper limit of normal the symptoms should be assumed to represent hepatic toxicity, and the patient should be evaluated as described below. The initial approach to gastrointestinal intolerance, not associated with hepatic toxicity, is to change the hour of drug administration and/or to administer the drugs with food. If patients are taking daily DOT, the timing of the drug administration should be altered, preferably to be closer to mealtime. Alternatively, food can be taken at the time of DOT administration. (In many programs food is offered as an incentive with DOT.) Patients receiving self-administered therapy can take the medications at bedtime. If gastrointestinal intolerance persists it may be best for all medications to be taken with meals. 6.3.2. Rash All drugs used in treating tuberculosis can cause a rash. The response to a patient with a rash depends on its severity. The rash may be minor, affecting a limited area or being predominantly manifested as itching, in which case antihistamines should be given for symptomatic relief, but all antituberculosis medications can be continued. A petechial rash may suggest thrombocytopenia in patients taking RIF (5). The platelet count should be checked and, if low, RIF hypersensitivity should be presumed to be the cause. RIF should be stopped and the platelet count monitored until it returns to baseline; RIF should not be restarted. If there is a generalized erythematous rash, especially if it is associated with fever and/or mucous membrane involvement, all drugs should be stopped immediately. If the patient has severe tuberculosis, three new drugs (e.g., an aminoglycoside and two oral agents) should be started. When the rash is substantially improved the medications can be restarted one by one, at intervals of 2--3 days. RIF should be restarted first (because it is the least likely to cause rash, and it is the most important agent), followed by INH, and then EMB or PZA. If the rash recurs the last drug added should be stopped. If no rash appears after the first three drugs have been restarted, the fourth drug should not be restarted unless the rash was relatively mild and the fourth drug is considered essential for therapy. 6.3.3. Drug fever Recurrence of fever in a patient who has been receiving therapy for several weeks should suggest drug fever, especially if the patient is showing microbiological and radiographic improvement. It should be noted, however, that fever from tuberculosis may persist for as long as 2 months after therapy has been initiated (6). Fever may also be a manifestation of a paradoxical reaction, especially in patients with HIV infection (see Section 8.1: HIV Infection) (7). The clinical hallmark of drug fever is that the patient looks and feels well despite having a high fever (often greater than 39ºC). There is no specific pattern to the fever. Eosinophilia may or may not be present. The first step in management of a possible drug fever is to ensure that there is no superinfection or worsening of tuberculosis. If these potential causes are excluded all drugs should be stopped. Drug-related fever usually will resolve within 24 hours. Patients with severe tuberculosis should be given at least three new drugs in the interim. Once the fever has resolved, the same protocol as described above for restarting drugs in the presence of a rash should be followed. 6.3.4. Hepatitis (Management of patients with baseline abnormal liver function is described in Section 8.8: Hepatic Disease.) Three of the first-line antituberculosis drugs, INH, RIF, and PZA, can cause drug-induced liver injury (AST level three or more times the upper limit of normal in the presence of symptoms, or five or more times the upper limit of normal in the absence of symptoms) (8). If the AST level is less than 5 times the upper limit of normal, toxicity can be considered mild, an AST level 5--10 times normal defines moderate toxicity, and an AST level greater than 10 times normal (i.e., greater than 500 IU) is severe (9). In addition to AST elevation, occasionally there are disproportionate increases in bilirubin and alkaline phosphatase. This pattern is more consistent with rifampin hepatotoxicity It is important to note that an asymptomatic increase in AST concentration occurs in nearly 20% of patients treated with the standard four-drug regimen (10). In the absence of symptoms therapy should not be altered because of modest asymptomatic elevations of AST, but the frequency of clinical and laboratory monitoring should be increased. In most patients, asymptomatic aminotransferase elevations resolve spontaneously. However, if AST levels are more than five times the upper limit of normal (with or without symptoms) or more than three times normal in the presence of symptoms, hepatotoxic drugs should be stopped immediately and the patient evaluated carefully. Similarly, a significant increase in bilirubin and/or alkaline phosphatase is cause for a prompt evaluation. Serologic testing for hepatitis A, B, and C should be performed and the patient questioned carefully regarding symptoms suggestive of biliary tract disease and exposures to other potential hepatotoxins, particularly alcohol and hepatotoxic medications. Drug-induced hepatitis is usually a diagnosis of exclusion but in view of the frequency with which other possible causes are present in any given patient, determining the cause may be difficult. Because the schedule for restarting antituberculosis medications is slower with hepatitis than for rash or drug fever it is generally prudent to give at least three nonhepatotoxic antituberculosis drugs until the specific cause of hepatotoxicity can be determined and an appropriate longer term regimen begun. The suspect antituberculosis medications should be restarted one at a time after the AST concentration returns to less than two times the upper limit of normal. (In patients with elevated baseline AST from preexisting liver disease, drugs should be restarted when the AST returns to near baseline levels.) Because RIF is much less likely to cause hepatotoxicity than is INH or PZA (Table 10) (10) and is the most effective agent, it should be restarted first. If there is no increase in AST after about 1 week, INH may be restarted. PZA can be started 1 week after INH if AST does not increase. If symptoms recur or AST increases the last drug added should be stopped. If RIF and INH are tolerated, and hepatitis was severe, PZA should be assumed to be responsible and should be discontinued. In this last circumstance, depending on the number of doses of PZA taken, severity of disease, and bacteriological status, therapy might be extended to 9 months. 6.4. Serum Drug Concentration Measurements The first-line drugs (INH, RIF, PZA, and EMB) have relatively predictable pharmacokinetics (11,12) and are highly efficacious when given in standard doses as DOT (13,14). Rarely, patients may have poor absorption or altered metabolism of the first-line drugs, resulting in failure of therapy (15,16) Second-line agents have a much narrower therapeutic window (the range of concentrations having reliable activity against M. tuberculosis but rarely causing toxicity) than the first-line drugs, and the consequences of treatment failure of drug-resistant tuberculosis may be difficult to manage. These considerations suggest several clinical situations in which therapeutic drug monitoring may be helpful: 1) patients with treatment failure that is not explained by nonadherence or drug resistance, 2) persons with medical conditions that may result in abnormal pharmacokinetics of the first-line drugs, and 3) the management of multidrug-resistant tuberculosis with second-line drugs. Be aware, however, that there are many uncertainties about the use of therapeutic drug monitoring in tuberculosis treatment. An important limitation is the lack of sufficient data to formulate clinically validated therapeutic ranges for antituberculosis agents. One response to the lack of clinically derived therapeutic ranges for the rifamycins is to use the distribution of concentrations achieved in healthy volunteers as the therapeutic range. However, in practice this approach has been quite problematic. For example, serum concentrations of the first-line drugs among HIV-infected patients with active tuberculosis are frequently lower than those in healthy volunteers (17,18), but HIV-related tuberculosis responds well to standard tuberculosis treatment regimens (19,20). The disadvantages of therapeutic drug monitoring are as follows: 1) the time necessary, from both patients and providers, to obtain and ship blood samples, and 2) the relatively high cost of measuring serum drug concentrations. Until more data are available, it seems prudent to restrict therapeutic drug monitoring for the first-line drugs to patients who are having an inadequate response to DOT (that is not due to nonadherence or drug resistance) or evidence of severe gastrointestinal or metabolic abnormalities. Examples of such circumstances include severe gastroparesis, short-bowel syndrome, chronic diarrhea with malabsorption, and renal insufficiency. As described above, patients with HIV-related tuberculosis may have an increased incidence of malabsorption of antituberculosis drugs (although some studies have contrary findings) (21,22). Even if true, this tendency for lower drug concentrations among patients with HIV-related tuberculosis is not sufficient to warrant routine therapeutic drug monitoring in this population. References
7. Drug Interactions7.1. Interactions Affecting Antituberculosis Drugs Drug--drug interactions can result in changes in the concentrations of one or both of the drugs involved. In the case of the antituberculosis drugs, there are relatively few interactions that substantially change the concentrations of the antituberculosis drugs; much more often the antituberculosis drugs cause clinically relevant changes in the concentrations of other drugs. The exceptions to this general rule are rifabutin and the fluoroquinolones. Rifabutin is partially metabolized by cytochrome P450 (CYP) 3A. Inhibitors of CYP3A increase serum concentrations of rifabutin and one of its metabolites (25-O-desacetyl-rifabutin), sometimes to toxic levels. For example, administration of ritonavir, a potent CYP3A inhibitor, with the standard daily dose of rifabutin (300 mg) increases the serum concentrations of rifabutin (4-fold increase) and 25-O-desacetyl-rifabutin (35-fold increase) (1) and is associated with increased rates of leukopenia, arthralgias, skin discoloration, and uveitis (2), all recognized to be toxic effects of rifabutin or one of its metabolites (3,4). Conversely, administering rifabutin with a CYP3A inducer decreases its concentrations, perhaps to ineffective levels. For example, efavirenz, a potent antiretroviral drug, decreases rifabutin serum concentrations by approximately one-third (5). Recommendations for making dose adjustments of rifabutin when it is given with commonly used CYP3A inhibitors and inducers are available (6,7). However, the complexity of these interactions and the rapidly changing nature of antiretroviral therapy strongly suggest that the management of cases of HIV-related tuberculosis should involve a physician with experience in this field. Absorption of the fluoroquinolones is markedly decreased by ingestion with medications containing divalent cations (calcium, iron, zinc), including antacids (8,9); supplements or vitamins containing calcium, iron or zinc (10), sucralfate (11); and the chewable tablet formulation of didanosine (12). These drug interactions can be avoided by assuring that medications containing divalent cations are ingested at least 2 hours apart from doses of fluoroquinolones (13). 7.2. Effects of Antituberculosis Drugs on Other Drugs 7.2.1. Drug interactions due to rifamycins The drugs used to treat tuberculosis affect the metabolism of many other drugs, and can result in a lack of efficacy (interactions with the rifamycins) or toxicity (interactions with isoniazid and the fluoroquinolones). Most of the clinically relevant drug--drug interactions involving the antituberculosis drugs are due to the effect of the rifamycins (rifampin, rifabutin, and rifapentine) on the metabolism of other drugs. All of the rifamycins are inducers of a variety of metabolic pathways, particularly those involving the various isozymes of the cytochrome P450 system (14--18). By inducing the activity of metabolic enzymes, rifamycin therapy results in a decrease in the serum concentrations of many drugs, sometimes to levels that are subtherapeutic. The rifamycins differ substantially in their potency as enzyme inducers; rifampin is the most potent, rifapentine is intermediate, and rifabutin is the least potent enzyme inducer (19). The well-described, clinically relevant drug--drug interactions involving the rifamycins are presented in Table 12 (1,5,15,20--88). However, it is important to note that many possible interactions involving the rifamycins have not been investigated fully and additional clinically relevant interactions undoubtedly will be described. Therefore, it is important to check all concomitant medications for possible, as well as confirmed, drug--drug interactions with rifamycins. Some of these drug--drug interactions can be managed with close clinical or laboratory monitoring and dose increases of the medication(s) affected by the rifamycins (Table 12). In other cases, the magnitude of the decrease in concentrations of a concomitant medication may be such that serum concentrations cannot be restored by a dose increase. If the dose of a medication is increased to compensate for the effect of a rifamycin, it is critical to remember that the dose of this drug will probably need to be decreased within the 2 weeks after the rifamycin is discontinued and its inductive effect resolves. In some situations, rifabutin can sometimes be used in place of rifampin, if there is an unacceptable drug--drug interaction between rifampin and another drug, such as cyclosporine (51) and most of the HIV-1 protease inhibitors (89). All the rifamycins may cause unacceptable decreases in the serum concentrations of certain drugs, such as delavirdine (26,27,90), ketoconazole and itraconazole (34,91). 7.2.2. Drug interactions due to isoniazid Isoniazid is a relatively potent inhibitor of several cytochrome P450 isozymes (CYP2C9, CYP2C19, and CYP2E1) (92), but has minimal effect on CYP3A (20). As an inhibitor, isoniazid can increase concentrations of some drugs to the point of toxicity. The clearest examples of toxicity due to the inhibitory activity of isoniazid are the anticonvulsants, phenytoin (93,94) and carbamazepine (95,96). Isoniazid also increases concentrations of benzodiazepines metabolized by oxidation, such as diazepam (85) and triazolam (97), but not those metabolized by conjugation, such as oxazepam (97). It is worth noting that rifampin has the opposite effect on the serum concentrations of many of these drugs. The available data demonstrate that the inductive effect of rifampin outweighs the inhibitory effect of isoniazid, so that the overall effect of combined therapy with rifampin and isoniazid is a decrease in the concentrations of drugs such as phenytoin (59) and diazepam (85). Isoniazid may increase toxicity of other drugs---acetaminophen (98), valproate (99), serotonergic antidepressants (100), disulfiram (101), warfarin (102), and theophylline (103)---but these potential interactions have not been well studied. 7.2.3. Drug interactions due to fluoroquinolones Ciprofloxacin (104) inhibits the metabolism of theophylline and can cause clinical theophylline toxicity (105). However, levofloxacin (106), gatifloxacin (107), and moxifloxacin (108) do not affect theophylline metabolism. References (Includes references cited in Table 12.)
8. Treatment in Special Situations8.1. HIV Infection Treatment of tuberculosis in patients with HIV infection follows the same principles as treatment of HIV-uninfected patients. However, there are several important differences between patients with and without HIV infection. These differences include the potential for drug interactions, especially between the rifamycins and antiretroviral agents, paradoxical reactions that may be interpreted as clinical worsening, and the potential for the development of acquired resistance to rifamycins when treated with highly intermittent therapy. 8.1.1. Clinical trials of treatment for tuberculosis in HIV-infected patients There have been seven prospective studies of 6-month regimens for the treatment of pulmonary tuberculosis in patients with HIV infection for which recurrence data were reported. Four of the studies were randomized, controlled trials (1--4), and three were observational in nature (5,6). These studies differed somewhat in design, patient population, eligibility criteria, frequency of dosing, treatment supervision, and outcome definitions; therefore, it is difficult to provide meaningful cross-study comparisons. All of the studies reported a good early clinical response to therapy and the time required for sputum culture conversion from positive to negative and treatment failure rates were similar to these indices of treatment efficacy in patients without HIV infection. Recurrence rates have varied among studies, with most reporting rates of 5% or less (2,3,5,6). In one study from the Democratic Republic of Congo (formerly Zaire), in which the recurrence rate in the 6-month arm was 9% compared with 3% in the 12-month arm, nonadherence in the continuation phase and/or exogenous reinfection may have contributed to the higher recurrence rate (1). In a randomized trial of once weekly INH-rifapentine versus twice weekly INH--RIF in the continuation phase of therapy, 5 of 30 (17%) HIV-infected patients receiving treatment in the once weekly arm relapsed compared with 3 of 31 (10%) patients in the twice weekly INH--RIF arm (4). Four of the five relapsed patients in the once weekly group had resistance to rifampin alone compared with none in the standard treatment arm. Because of the small sample size in the standard treatment arm, it is difficult to interpret the relapse rate of 10%. In an observational study of twice weekly INH--rifabutin among HIV-infected tuberculosis patients also receiving antiretroviral therapy, 7 of 156 patients failed treatment or relapsed (7). Although the life table rate of failure/relapse was low (4.6%), M. tuberculosis isolated from all five of these patients was resistant to RIF alone. The phenomenon of acquired rifampin monoresistance was also seen in a trial of largely twice weekly INH--RIF therapy, albeit at a lower rate (3). In all of these studies, acquired RIF resistance occurred only among patients with CD4+ cell counts <100 cells/µl. Acquired rifampin resistance has not been seen in trials where RIF was given daily. A consistent finding in the treatment studies has been a high mortality rate among HIV-seropositive patients. In most studies the cause of death is difficult to ascertain. Early mortality may be related to advanced tuberculosis, but deaths during the continuation phase of therapy are usually due to other AIDS-related conditions. Mortality during treatment among HIV-infected patients with tuberculosis has been associated with advanced HIV disease (1,3,6,8). However, the use of effective antiretroviral therapy during the treatment of tuberculosis in persons with HIV infection may improve treatment outcomes and, thus, is recommended, as described subsequently (9). A major concern in treating tuberculosis in the setting of HIV infection is the interaction of RIF with antiretroviral agents (see Section 7: Drug Interactions, and Table 12). As described previously, rifabutin is highly active against M. tuberculosis but has less of an effect in inducing hepatic microsomal enzymes than RIF. Data from clinical trials suggest that rifabutin and RIF-based regimens are equally efficacious. Gonzalez-Montaner and colleagues (10) reported the first randomized clinical trial comparing rifabutin (150 and 300 mg) with RIF in a 6-month regimen in persons without HIV infection. The outcomes were highly favorable in both groups and there were few adverse reactions. Investigators from South Africa reported a randomized, open-label trial comparing rifabutin with RIF in a standard four-drug regimen administered with DOT (11). Although patients did not have HIV testing performed, the HIV seroprevalence was reportedly low at the time of the study. In the continuation phase, the medications were given twice weekly. By 2 months after treatment was begun, 88% of the patients in the RIF arm and 92% of those given rifabutin had negative sputum cultures. The relapse rate was 3.8% in the RIF group versus 5.1% in the rifabutin group (p = NS). Only one study examining the effectiveness of rifabutin included HIV-infected patients (12). A single blind randomized study of 50 HIV-infected patients in Uganda compared a fully supervised regimen of RIF versus rifabutin together with INH, EMB, and PZA. Time to sputum conversion was similar between groups when controlling for baseline characteristics. Relapse data were not available. Investigators in Uganda have reported a higher mortality rate among HIV-infected patients treated with regimens that did not contain RIF. Wallis and associates (13) reported that a non-RIF-containing regimen was associated with shortened survival compared with an RIF-based regimen. In addition to the higher mortality associated with non-RIF-based regimens, other studies have demonstrated unacceptably high recurrence rates in the setting of HIV infection (14,15). Thus, every effort should be made to use a rifamycin-based regimen for the entire course of therapy in persons with HIV infection. 8.1.2. Treatment recommendations Recommendations for the treatment of tuberculosis in HIV-infected adults are, with two exceptions, identical to those for HIV-uninfected adults: a 6-month regimen consisting of an initial phase of INH, RIF, PZA, and EMB given for 2 months followed by INH and RIF for 4 months when the disease is caused by organisms that are known or presumed to be susceptible to the first-line agents. This regimen may be given by daily or intermittent administration as listed in Table 1 and described in Section 5.2: Recommended Regimens. However, on the basis of data showing an increased frequency of rifamycin resistance among patients having CD4+ cell counts <100/µl, it is recommended that patients with advanced HIV disease be treated with daily or three times weekly therapy in the continuation phase (Rating AIII) (16). Twice weekly drug administration in the continuation phase should not be used in patients with CD4+ cell counts <100/µl. Twice weekly therapy may be considered in patients with less advanced immunosuppression (CD4+ cell counts >100/µl). Once weekly administration of INH--rifapentine in the continuation phase should not be used in any patient with HIV infection. Six months should be considered the minimum duration of treatment for adults, even for patients with culture-negative tuberculosis. If there is evidence of a slow or suboptimal response (e.g., cultures are still positive after 2 months of therapy), prolongation of the continuation phase to 7 months (a total of 9 months treatment) should be strongly considered. DOT and other adherence-promoting strategies should be used in all patients with HIV-related tuberculosis. Although there are no data on which to base recommendations, the American Academy of Pediatrics recommends that for HIV-infected children the minimum duration of therapy be 9 months (17). All patients with tuberculosis should be advised to undergo voluntary counseling and HIV testing. Efforts should be made to engage all patients with a new diagnosis of HIV infection in HIV care during their treatment for tuberculosis. Ideally, patients should be managed by physicians who are expert in the treatment of tuberculosis/HIV coinfection. If the HIV care provider and tuberculosis care provider are not the same person, communication between them is essential and should occur frequently throughout the course of treatment. 8.1.3. Safety and tolerability The frequency of antituberculosis drug-related toxicity in patients with HIV infection has varied from study to study. In a retrospective study from San Francisco, 18% of HIV-seropositive patients with tuberculosis had a change of regimen because of adverse drug reactions (18). RIF was the drug implicated most commonly, producing an adverse reaction in 12% of the patients. In the Democratic Republic of Congo, 11% of the seropositive patients developed a rash but in none was the treatment interrupted (1). Paresthesia developed in 21% of the cases, suggesting the need for pyridoxine when treating tuberculosis in persons with HIV infection. Other investigators have reported low rates of significant adverse reactions (3,5,6,19). In the three times weekly regimen studied in Haiti, there were no differences in adverse events between HIV-infected and uninfected patients (6). In HIV-infected patients it is often difficult to distinguish an adverse reaction to antituberculosis drugs from the effects of associated conditions or reactions to any of the many medications that are often being taken concurrently. Because of the difficulties in diagnosing a drug reaction and in determining the responsible agent, the first-line antituberculosis drugs (especially INH or RIF) should not be stopped permanently without strong evidence that the antituberculosis drug was the cause of the reaction. In such situations consultation with an expert in treating tuberculosis in persons with HIV infection is recommended. In a study reported by Ungo and associates (20), it was demonstrated that the relative risk of developing drug-induced hepatoxicity in tuberculosis patients with hepatitis C virus or HIV infection was 5- and 4-fold, respectively, compared with a 14-fold relative risk in patients with both hepatitis C virus and HIV infections. This finding was not confirmed in a study from Baltimore, in which rates of transaminase elevation were not greater in patients with HIV and hepatitis C virus who were given INH (21). Current IDSA and USPHS guidelines recommend screening all HIV-infected patients for hepatitis C virus (22). Until more data are available it is probably prudent to provide more frequent clinical and laboratory monitoring, as described for patients with preexisting liver disease, for patients with HIV infection or hepatitis C virus infection who are being treated for tuberculosis. 8.1.4. Concurrent administration of antiretroviral agents and rifamycins Most patients with tuberculosis have relatively advanced HIV disease and, thus, antiretroviral therapy is indicated (23). Antiretroviral therapy should not be withheld simply because the patient is being treated for tuberculosis, if it is otherwise indicated. Nevertheless, it is not advisable to begin both antiretroviral therapy and combination chemotherapy for tuberculosis at nearly the same time. So doing may involve as many as eight new drugs with interactions and overlapping toxicities that would be difficult to evaluate. Although there are few data on which to base recommendations, expert opinion suggests that treatment for tuberculosis should be initiated first. Although antiretroviral therapy has a dramatic effect in decreasing progression of HIV disease (decreasing CD4+ cell counts, new opportunistic infections, or death), among patients with HIV-related tuberculosis, the use of antiretroviral therapy in the setting of tuberculosis therapy is complex. In those patients not already receiving antiretroviral therapy, early initiation of antiretroviral therapy may decrease HIV disease progression, but is also associated with a high incidence of side effects and paradoxical reactions, some severe enough to warrant discontinuation of both antiretroviral and antituberculosis drugs (9). In addition, starting so many new medications in a short time period may present a tremendous adherence challenge for patients adjusting to the diagnoses of both tuberculosis and AIDS. Delaying the initiation of antiretroviral therapy until 4--8 weeks after starting antituberculosis therapy has the potential advantages of being better able to ascribe a specific cause for a drug side effect, decreasing the severity of paradoxical reactions, and decreasing the adherence difficulties for the patient. Until there have been controlled studies evaluating the optimal time for starting antiretroviral therapy in patients with HIV infection and tuberculosis, this decision should be individualized, based on the patient's initial response to treatment for tuberculosis, occurrence of side effects, and ready availability of multidrug antiretroviral therapy. For patients with CD4+ cell counts >350 cells/µl, the antiretroviral regimen could be initiated at any time after tuberculosis treatment was begun, based on current recommendations (23). For patients who are already receiving an antiretroviral regimen, treatment should generally be continued, although the regimen may need to be modified on the basis of the risk of drug--drug interactions, as described in Section 7: Drug Interactions. Even though drug interactions are common, a rifamycin should not be excluded from the tuberculosis treatment regimen for fear of interactions with some antiretroviral agents. The exclusion of a rifamycin from the treatment regimen is likely to delay sputum conversion, will prolong the duration of therapy, and possibly result in a poorer outcome (24). As noted in Section 7, Drug Interactions, rifabutin has fewer interactions than RIF and should be used if these categories of antiretroviral agents are being administered. The categories of antiretroviral agents available currently are nucleoside reverse transcriptase inhibitors (NRTIs), nucleotide reverse transcriptase inhibitors (NtRTIs), nonnucleoside reverse transcriptase inhibitors (NNRTIs), and protease inhibitors (PIs). The NRTIs and NtRTIs do not have clinically significant drug interactions with the standard antituberculosis medications; thus, drugs in these categories can be used together with rifamycins without any dose adjustment being necessary. However, the PIs and NNRTIs, depending on the specific drug, may either inhibit or induce cytochrome P450 isoenzymes (CYP450). Thus, these drugs may alter the serum concentration of rifabutin, as described in Section 7.1: Interactions Affecting Antituberculosis Drugs. When rifabutin is combined with antiretroviral agents, its dose and the dose of the antiretroviral agents may require adjustment. A report described the successful use of rifabutin with an antiretroviral regimen containing PIs (25). All 25 patients became culture negative by 2 months and no relapses were reported after a median follow-up of 13 months. Moreover, the circulating HIV RNA levels decreased significantly, with 20 of 25 patients achieving viral loads of less than 500 copies/ml. Thus, it appears that both tuberculosis and HIV can be treated successfully with concurrent use of a rifabutin-based regimen and potent combinations of antiretroviral agents. Previous guidelines from CDC specifically stated that RIF was contraindicated in patients who were taking any PI or NNRTI (26). However, new data indicate that RIF can be used for the treatment of tuberculosis with certain combinations of antiretroviral agents (27,28). As recommended by CDC (27), rifampin can be used with a regimen of efavirenz and two NRTIs, with ritonavir and one or more NRTIs, with ritonavir and saquinavir (either hard-gel or soft-gel capsule), and with a triple nucleoside regimen. As new antiretroviral agents and more pharmacokinetic data become available, these recommendations are likely to be modified. Because these recommendations are frequently revised, obtaining the most up-to-date information from the CDC website, http://www.cdc.gov/nchstp/tb/, is advised. Updated information on antiretroviral drugs and drug interactions, compiled by Medscape, can be found at http://www.medscape.com/updates/quickguide. When starting NNRTIs or PIs for tuberculosis patients receiving RIF, a 2-week "washout" period is generally recommended between the last dose of RIF and the first dose of PIs or NNRTIs to allow for reduction of the enzyme-inducing activity of RIF. During this time, rifabutin may be started to ensure that the tuberculosis treatment regimen is adequate. For patients already receiving antiretroviral agents at the time treatment for tuberculosis is begun, an assessment of the antiretroviral regimen should be undertaken and, if necessary, changes made to ensure optimum treatment of the HIV infection during tuberculosis therapy. Conversely, the determination of whether to use RIF and the dose of the rifamycin must take into account the antiretroviral regimen. 8.1.5. Paradoxical reaction On occasion, patients have a temporary exacerbation of symptoms, signs, or radiographic manifestations of tuberculosis (paradoxical reaction) after beginning antituberculosis treatment. Worsening of this sort occurs in patients without HIV infection, especially with lymphadenitis, but it is more common among HIV-infected patients. These reactions presumably develop as a consequence of reconstitution of immune responsiveness brought about by antiretroviral therapy or, perhaps, by treatment of the tuberculosis itself. Narita and colleagues (29) reported that among HIV-infected patients who were taking antiretroviral agents, 36% developed paradoxical worsening after beginning treatment for tuberculosis compared with 7% of those who were not taking antiretroviral drugs. In contrast, Wendel and colleagues (30) reported that only 7% of HIV-infected patients with tuberculosis developed paradoxical worsening and the reactions were not associated with antiretroviral therapy. Signs of a paradoxical reaction may include high fevers, increase in size and inflammation of involved lymph nodes, new lymphadenopathy, expanding central nervous system lesions, worsening of pulmonary parenchymal infiltrations, and increasing pleural effusions. Such findings should be attributed to a paradoxical reaction only after a thorough evaluation has excluded other possible causes, especially tuberculosis treatment failure. A paradoxical reaction that is not severe should be treated symptomatically without a change in antituberculosis or antiretroviral therapy. Although approaches to the management of severe reactions, such as high fever, airway compromise from enlarging lymph nodes, enlarging serosal fluid collections, and sepsis syndrome, have not been studied, expert opinion suggests that prednisone or methylprednisolone be started at a dose of about 1 mg/kg and gradually reduced after 1 to 2 weeks. References
 8.2. Children and Adolescents Children most commonly develop tuberculosis as a complication of the initial infection with M. tuberculosis (primary tuberculosis). Radiographically, primary tuberculosis is characterized by intrathoracic adenopathy, mid- and lower lung zone infiltrates, and the absence of cavitation. However, children, occasionally, and adolescents, more frequently, develop adult-type tuberculosis (upper lobe infiltration and cavitation associated with sputum production). The lesions of primary tuberculosis have a smaller number of M. tuberculosis organisms than those of adult-type pulmonary tuberculosis; thus, treatment failure, relapse, and development of secondary resistance are rare phenomena among children. Because it is more difficult to isolate M. tuberculosis from a child with pulmonary tuberculosis than from an adult, it is frequently necessary to rely on the results of culture and susceptibility tests of specimens from the person presumed to be the source of the infection in the child to guide the choice of drugs for the child. In children in whom drug resistance is suspected or for whom no source case isolate is available, attempts to isolate organisms via three early morning gastric aspirations (optimally during hospitalization), bronchoalveolar lavage, or tissue biopsy must be considered. Because tuberculosis in infants and children younger than 4 years of age is more likely to disseminate, treatment should be started as soon as the diagnosis is suspected. Asymptomatic children with a positive PPD-tuberculin skin test and an abnormal chest radiograph (atelectasis, parenchymal infiltrate, or hilar adenopathy) should receive combination chemotherapy, usually with INH, RIF, and PZA as initial therapy. Several controlled and observational trials of 6-month therapy in children with pulmonary tuberculosis caused by organisms known or presumed to be susceptible to the first-line drugs have been published (1--9). Six months of therapy with INH and RIF has been shown to be effective for hilar adenopathy and pulmonary disease caused by drug-susceptible organisms (5,6). However, most studies used 6 months of daily treatment with INH and RIF, supplemented during the first 2 weeks to 2 months with PZA. This three-drug combination has a success rate of greater than 95% and a rate of adverse effects of less than 2%. Two studies used twice or three times weekly therapy from the beginning with good results (1,7). Many experts prefer to treat children with three (rather than four) drugs in the initial phase because the bacillary population is low, because many infants and children cannot tolerate the pill burden required with four oral drugs, and because of the difficulty in performing visual acuity tests in young children who are being treated with EMB. In children suspected or known to have been infected with an M. tuberculosis strain that is fully susceptible, the initial phase should consist of INH, RIF, and PZA. If the susceptibility of the presumed infecting strain is not known and the likelihood of failure is low (primary tuberculosis), some experts prefer to use three drugs. However, children and adolescents with adult-type pulmonary tuberculosis, as defined above, should be treated with the four-drug initial phase regimen, unless the infecting strain is known to be susceptible (10). When epidemiologic circumstances (Table 6) suggest an increased risk of drug-resistant organisms being present, EMB can be used safely in a dose of about 15--20 mg/kg per day, even in children too young for routine eye testing. Older children should have monthly evaluations of visual acuity and color discrimination while taking EMB. SM, kanamycin, or amikacin can be used as the fourth drug, when necessary. The usual doses for daily and twice weekly treatment in children are listed in Section 3, Drugs in Current Use, and shown in Table 3. Three times weekly therapy is not recommended for children. Pyridoxine is recommended for infants, children, and adolescents who are being treated with INH and who have nutritional deficiencies, symptomatic HIV infection, or who are breastfeeding. DOT should be used for all children with tuberculosis. The lack of pediatric dosage forms of most antituberculosis medications necessitates using crushed pills and suspensions. Even when drugs are given under DOT, tolerance of the medications must be monitored closely. Parents should not be relied on to supervise DOT. Because of the difficulties in isolating M. tuberculosis from children, bacteriological examinations are less useful in evaluating the response to treatment and clinical and radiographic examinations are of relatively greater importance. However, hilar adenopathy and resultant atelectasis may require 2--3 years to resolve. Thus, a persisting abnormality on chest radiographs is not necessarily a criterion for extending continuing therapy. Recognition of treatment failure or relapse in a child is subject to the same difficulties as making a diagnosis. Thus, clinical and radiographic worsening may not be accompanied by positive AFB smears or mycobacterial cultures. A decision to modify the drug regimen should not be made lightly, but often must be made on clinical grounds only. In general, extrapulmonary tuberculosis in children can be treated with the same regimens as pulmonary disease. Exceptions may be disseminated disease, and meningitis, for which there are inadequate data to support 6-month therapy. A fourth drug is recommended in the initial phase when there is disseminated tuberculosis. The recommended duration is 9--12 months. The optimal treatment of pulmonary tuberculosis in children and adolescents with HIV infection is unknown. The American Academy of Pediatrics recommends that initial therapy should always include at least three drugs (INH and RIF, plus PZA for the first 2 months), and the total duration of therapy should be at least 9 months (11). References
8.3. Extrapulmonary Tuberculosis Tuberculosis can involve virtually any organ or tissue in the body. Nonpulmonary sites tend to be more common among children and persons with impaired immunity. To establish the diagnosis of extrapulmonary tuberculosis, appropriate specimens including pleural fluid; pericardial or peritoneal fluid; pleural, pericardial, and peritoneal biopsy specimens; lymph node tissue; and bone marrow, bone, blood, urine, brain, or cerebrospinal fluid should be obtained for AFB staining, mycobacterial culture, and drug susceptibility testing (1). Tissue specimens should also be examined microscopically, after routine and AFB staining, but the absence of AFB and of granulomas or even failure to culture M. tuberculosis does not exclude the diagnosis of tuberculosis. Bacteriological evaluation of the response to treatment in extrapulmonary tuberculosis is often limited by the difficulty in obtaining follow-up specimens. Thus, response often must be judged on the basis of clinical and radiographic findings. The basic principles that underlie the treatment of pulmonary tuberculosis also apply to extrapulmonary forms of the disease. Although many fewer treatment studies have examined treatment of extrapulmonary tuberculosis, compared with pulmonary disease, increasing evidence, including some randomized controlled trials, suggests that 6- to 9-month regimens that include INH and RIF are effective (2--16). Therefore, among patients with extrapulmonary tuberculosis, a 6- to 9-month regimen (2 months of INH, RIF, PZA, and EMB followed by 4--7 months of INH and RIF) is recommended as initial therapy unless the organisms are known or strongly suspected of being resistant to the first-line drugs. If PZA cannot be used in the initial phase, the continuation phase must be increased to 7 months, as described for pulmonary tuberculosis. The exception to the recommendation for a 6- to 9-month regimen is tuberculous meningitis, for which the optimal length of therapy has not been established, but some experts recommend 9--12 months. Although in extrapulmonary tuberculosis there have not been controlled trials of the various patterns of intermittent drug administration listed in Table 2, expert opinion suggests that all could be used, with the exception of INH--rifapentine once weekly in the continuation phase. Given the lack of experience with this regimen, it is not recommended currently for treating extrapulmonary tuberculosis. Corticosteroid treatment is a useful adjunct in treating some forms of extrapulmonary tuberculosis, specifically meningitis and pericarditis caused by drug-susceptible organisms. Evidence-based recommendations on the duration of treatment for extrapulmonary tuberculosis and the use of corticosteriods are shown in Table 13. 8.3.1. Lymph node tuberculosis A 6-month regimen as described in Section 5, Recommended Treatment Regimens, and Table 2 is recommended for initial treatment of all patients with tuberculous lymphadenitis caused by drug-susceptible organisms (2--6). Affected lymph nodes may enlarge while patients are receiving appropriate therapy or after the end of treatment without any evidence of bacteriological relapse (3,5,17,18). On occasion, new nodes can appear during or after treatment as well. Therapeutic lymph node excision is not indicated except in unusual circumstances. For large lymph nodes that are fluctuant and appear to be about to drain spontaneously, aspiration or incision and drainage appears to be beneficial, although this approach has not been examined systematically (Rating BIII). It should be noted that the majority of cases of lymphatic mycobacterial disease in children born in the United States are caused by nontuberculous mycobacteria. 8.3.2. Bone and joint tuberculosis Several studies have examined treatment of bone and joint tuberculosis and have shown that 6- to 9-month regimens containing RIF are at least as effective as 18-month regimens that do not contain RIF (13--15) Because of the difficulties in assessing response, however, some experts tend to favor the 9-month duration. A randomized trial performed primarily among ambulatory patients by the Medical Research Council Working Party on Tuberculosis of the Spine (13) demonstrated no additional benefit of surgical debridement or radical operation (resection of the spinal focus and bone grafting) in combination with chemotherapy compared with chemotherapy alone. Myelopathy with or without functional impairment most often responds to chemotherapy. In two Medical Research Council studies conducted in Korea, 24 of 30 patients in one study (14) and 74 of 85 patients in an earlier study (19) had complete resolution of myelopathy or complete functional recovery when treated medically. In some circumstances, however, surgery appears to be beneficial and may be indicated. Such situations include failure to respond to chemotherapy with evidence of ongoing infection, the relief of cord compression in patients with persistence or recurrence of neurologic deficits, or instability of the spine. 8.3.3. Pericardial tuberculosis For patients with pericardial tuberculosis, a 6-month regimen is recommended. Corticosteroids are recommended as adjunctive therapy for tuberculous pericarditis during the first 11 weeks of antituberculosis therapy. In a randomized, double-blind, controlled trial, patients in the later effusive--constrictive phase who received prednisolone had a significantly more rapid clinical resolution compared with patients given placebo. Prednisolone-treated patients also had a lower mortality (2 of 53 [4%] versus 7 of 61 [11%]) and needed pericardiectomy less frequently (11 of 53 [21%] versus 18 of 61 [30%]), but the differences did not reach statistical significance (8). Prednisolone did not reduce the risk of constrictive pericarditis. In a second prospective, double-blind, randomized trial of adjunctive prednisolone therapy involving patients with effusive pericarditis (i.e., more acute disease), prednisolone reduced the need for repeated pericardiocentesis (7 of 76 [9%] versus 17 of 74 [23%]; p <0.05) and was associated with a significantly lower mortality (2 of 76 [3%] died among those who received prednisolone compared with 10 of 74 [14%] among those not given prednisolone; p <0.05) (9). As before, there was no statistically significant impact on progression to constriction or in the need for pericardiectomy. An additional small randomized trial by Hakim and associates (20) performed in HIV-infected patients with tuberculous pericarditis also demonstrated that prednisolone therapy was associated with a reduced risk of mortality. On the basis of these studies, it is recommended that daily adjunctive prednisolone or prednisone treatment be given to adults and children with tuberculous pericarditis. For adults the prednisone dose is 60 mg/day (or the equivalent dose of prednisolone) given for 4 weeks, followed by 30 mg/day for 4 weeks, 15 mg/day for 2 weeks, and finally 5 mg/day for week 11 (the final week). Children should be treated with doses proportionate to their weight, beginning with about 1 mg/kg body weight and decreasing the dose as described for adults. 8.3.4. Pleural tuberculosis A 6-month regimen is also recommended for treating pleural tuberculosis. A number of studies have examined the role of corticosteroid therapy for tuberculous pleural effusions (21), but only two have been prospective, double blind, and randomized (7,22). In both of these studies, prednisone (or prednisolone) administration did not reduce the development of residual pleural thickening. Lee and associates (22) found that patients with pleural tuberculosis who received prednisone had a significantly more rapid resolution of symptoms such as fever, chest pain, and dyspnea than patients given placebo. Patients who received prednisone had a more rapid radiographic resolution of the effusions. In the study by Wyser and colleagues (7), all patients had complete drainage of the effusion performed at the time of the diagnostic procedure; patients were then allocated at random to receive adjunctive oral prednisone or placebo for 6 weeks. The complete drainage led to a rapid resolution of symptoms, and the added benefit of corticosteroids on symptoms was minimal. Tuberculous empyema, a chronic, active infection of the pleural space containing a large number of tubercle bacilli, usually occurs when a cavity ruptures into the pleural space. Treatment consists of drainage (often requiring a surgical procedure) and antituberculous chemotherapy. Surgery, when needed, should be undertaken by experienced thoracic surgeons (23). The optimum duration of treatment for this unusual form of tuberculosis has not been established. 8.3.5. Tuberculous meningitis Before the advent of effective antituberculosis chemotherapy, tuberculous meningitis was uniformly fatal. Tuberculous meningitis remains a potentially devastating disease that is associated with a high morbidity and mortality, despite prompt initiation of adequate chemotherapy (24--29). HIV-infected patients appear to be at increased risk for developing tuberculous meningitis but the clinical features and outcomes of the disease are similar to those in patients without HIV infection (24--26,29). Patients presenting with more severe neurologic impairment such as drowsiness, obtundation, or coma have a greater risk of neurologic sequelae and a higher mortality. Chemotherapy should be initiated with INH, RIF, PZA, and EMB in an initial 2-month phase. INH and RIF, as well as the aminoglycosides, capreomycin, and the fluoroquinolones are available in parenteral forms for patients with altered mental status who may not be able to take oral medications. After 2 months of four-drug therapy for meningitis caused by susceptible strains, PZA and EMB may be discontinued, and INH and RIF continued for an additional 7--10 months, although the optimal duration of chemotherapy is not defined, and there are no data from randomized, controlled trials to serve as the basis of recommendations. Repeated lumbar punctures should be considered to monitor changes in CSF cell count, glucose, and protein, especially in the early course of therapy. Differences in regimens among patient groups and in the use of corticosteroid therapy have made meta-analysis of published treatment trials impossible (30). Some authors have advocated longer courses of therapy, up to 2 years (28,31), whereas others have suggested that short-course RIF-based regimens for 6 to 9 months may be adequate therapy (10,32,33). It has been reported that some patients being treated for tuberculous meningitis develop tuberculomas during therapy, perhaps as a form of paradoxical reaction; however, this does not necessarily indicate treatment failure. A number of investigators have examined the role of adjunctive corticosteroid therapy in the treatment of tuberculous meningitis (21,34--41), but many of these are limited by small sample size or use of a regimen that did not include RIF. There are no large, prospective, randomized, controlled trials of adjunctive corticosteroid use for tuberculous meningitis in which an RIF-based regimen has been used. Six of eight controlled trials noted a benefit of corticosteroid therapy in terms of survival, frequency of sequelae, or both. In the study conducted by Girgis and coworkers (34), the greatest benefit was for patients with Stage II disease (lethargic) on presentation (4 of 27 [15%] of those who received dexamethasone died versus 14 of 35 [40%] in the control group; p <0.02). For patients presenting with coma (Stage III), there was no significant difference in survival between those who received dexamethasone and control patients (28 of 44 [64%] mortality for the dexamethasone group versus 35 of 46 [76%] for control subjects). However, the small sample size may have precluded finding an effect. Likewise, there were too few patients with Stage I disease (alert) on entry to determine the effectiveness of dexamethasone for this less severely ill group. On the basis of the available data, albeit limited, adjunctive corticosteroid therapy with dexamethasone is recommended for all patients, particularly those with a decreased level of consciousness, with tuberculous meningitis. The recommended regimen is dexamethasone in an initial dose of 8 mg/day for children weighing less than 25 kg and 12 mg/day for children weighing 25 kg or more and for adults. The initial dose is given for 3 weeks and then decreased gradually during the following 3 weeks. 8.3.6. Disseminated tuberculosis A 6-month regimen is recommended for tuberculosis at multiple sites and for miliary tuberculosis, although there are limited data from controlled clinical trials addressing this issue. (The AAP recommends 9 months of treatment for children with disseminated tuberculosis.) Expert opinion suggests that corticosteroid therapy may be useful for treating respiratory failure caused by disseminated tuberculosis but there are no data to support its use. 8.3.7. Genitourinary tuberculosis Renal tuberculosis is treated primarily with medical therapy (12,42--46), and a 6-month regimen is recommended. If ureteral obstruction occurs, procedures to relieve the obstruction are indicated. In cases of hydronephrosis and progressive renal insufficiency due to obstruction, renal drainage by stenting or nephrostomy is recommended (42). The use of corticosteriods in addition to stenting for the treatment of ureteric stenosis is discussed in the urologic literature but the efficacy of steroids in this setting is unclear. Nephrectomy is not usually indicated for the treatment of uncomplicated renal tuberculosis but should be considered when there is a nonfunctioning or poorly functioning kidney, particularly if hypertension or continuous flank pain is present. Tuberculosis of either the female or male genital tract responds well to standard chemotherapy, and surgery is needed only for residual large tubo-ovarian abscesses. A positive urine culture for M. tuberculosis occurs relatively commonly as an incidental finding among patients with pulmonary or disseminated disease, especially those with HIV infection. The positive culture may occur in the absence of any abnormalities on urinalysis and does not necessarily represent genitourinary tract involvement. 8.3.8. Abdominal tuberculosis A 6-month regimen is recommended for patients with peritoneal or intestinal tuberculosis (47,48). There are insufficient data to recommend adjunctive corticosteroid therapy in the treatment of tuberculous peritonitis (21). In a small study of peritoneal tuberculosis alternate patients received adjunctive corticosteroid therapy for 4 months (total of 23 steroid recipients) (49). Fibrotic complications were noted in 4 of 24 in the control group and in none of those in the steroid group (23 patients), but the difference was not statistically significant. 8.3.9. Other sites of involvement As noted above, tuberculosis can involve any organ or tissue. In treating tuberculosis in sites other than those mentioned, the basic principles of therapy apply, but experts should be consulted for specific advice concerning individual patients. References
8.4. Culture-Negative Pulmonary Tuberculosis in Adults Failure to isolate M. tuberculosis from appropriately collected specimens in persons who, because of clinical or radiographic findings, are suspected of having pulmonary tuberculosis does not exclude a diagnosis of active tuberculosis. For the United States as a whole, about 17% of the reported new cases of pulmonary tuberculosis have negative cultures (1). Low bacillary populations, temporal variations in the number of bacilli being expelled, and errors in specimen processing all may result in failure to isolate organisms from patients who have active tuberculosis. It should be emphasized that alternative diagnoses must be considered carefully and appropriate diagnostic studies undertaken in patients who have what appears to be culture-negative tuberculosis. At a minimum, patients suspected of having pulmonary tuberculosis should have three sputum specimens (using sputum induction with hypertonic saline if necessary) for AFB smears and cultures for mycobacteria as part of the diagnostic evaluation. Depending on the clinical features and differential diagnosis, other diagnostic testing, such as bronchoscopy with bronchoalveolar lavage and biopsy, should be considered before making a presumptive diagnosis of culture-negative tuberculosis. Patients who, on the basis of careful clinical and radiographic evaluation, are thought to have pulmonary tuberculosis should have treatment initiated with INH, RIF, PZA, and EMB even when the initial sputum smears are negative. If M. tuberculosis is isolated in culture, treatment for active disease should be continued. Patients who have negative cultures but who still are presumed to have pulmonary tuberculosis should have a thorough follow-up clinical and radiographic evaluation at the time 2 months of therapy has been completed to determine whether there has been a response that can be attributed to antituberculosis treatment. If there is either clinical or radiographic improvement and no other etiology is identified, treatment should be continued for active tuberculosis. A 4-month, INH and RIF regimen for culture-negative tuberculosis has been demonstrated to be successful with only 1.2% relapses during an average follow-up of 44 months (2). However, because the results of cultures may not be known for 3--8 weeks and because of the possibility of drug resistance, initiation of two-drug therapy with INH and RIF alone is not recommended, but the continuation phase can be shortened to 2 months using INH and RIF (Figure 2). On occasion, patients who are being evaluated for pulmonary tuberculosis will be found to have positive AFB smears but negative cultures. There are several potential explanations for this occurrence, including the possibilities that the acid-fast organisms are nontuberculous and difficult to culture, that they are nonviable tubercle bacilli, and that they are the result of laboratory error. The approach taken in such cases should be individualized on the basis of clinical and radiographic findings. If suspicion of tuberculosis is high and the patient has positive AFB smears, even with negative cultures, he/she should be treated as if the culture is positive, using one of the recommended regimens. References
8.5. Radiographic Evidence of Prior Tuberculosis: Inactive Tuberculosis Persons with a positive tuberculin PPD skin test who have radiographic findings consistent with prior pulmonary tuberculosis (ATS/CDC Class 4) (1) and who have not been treated are at increased risk for the subsequent development of active tuberculosis (2--4). The radiographic findings that constitute evidence of prior tuberculosis are apical fibronodular infiltrations, often with volume loss. Case rates among such persons in one study were about 2.5 times those of persons infected with M. tuberculosis who did not have chest radiographic abnormalities (3). Persons with radiographic findings of healed primary tuberculosis (e.g., calcified solitary pulmonary nodules, calcified hilar lymph nodes, and pleural thickening) are not at increased risk for tuberculosis compared with other persons with latent tuberculosis infection. Patients should not be classified as having radiographic evidence of prior tuberculosis if another disease is found to account for the radiographic findings. The activity of tuberculosis cannot be determined from a single chest radiograph, and unless there are previous radiographs showing that the abnormality has not changed, it is recommended that sputum examination, using sputum induction if necessary, be performed to assess the possibility of active tuberculosis. Once active tuberculosis has been excluded by sputum culture, these persons are high-priority candidates for treatment of latent tuberculosis infection (5). The optimum treatment for patients with latent tuberculosis infection and abnormal chest radiographs consistent with prior tuberculosis has been examined in several studies. A placebo-controlled trial conducted by the IUATLD (2) compared the efficacy of 3, 6, and 12 months of INH in preventing active tuberculosis for persons with latent tuberculosis infection who had chest radiographs showing fibrotic lesions consistent with inactive tuberculosis. Among those receiving INH for at least 6 months, the incidence of tuberculosis was significantly reduced compared with those given placebo. In patients with fibrotic lesions greater than 2 cm in diameter INH given for 12 months was significantly better than 6 months (89 versus 67% reduction). A reanalysis of data from a community-based study of persons with abnormal radiographs felt to represent inactive tuberculosis showed that the efficacy of INH decreased significantly if less than 9 months of the drug was taken, but that further protection was not conferred if the duration was extended from 9 to 12 months (6). On the basis of these data, guidelines for treatment of latent tuberculosis infection recommend 9 months of INH for persons with abnormal chest radiographs consistent with prior tuberculosis (5). Additional treatment regimens are RIF (with or without INH) for 4 months, and RIF and PZA for 2 months (for persons who are unlikely to complete a longer course and who can be monitored carefully) (5) (Table 14). A study comparing the cost-effectiveness of INH and RIF with INH alone in treating this category of patient showed that 4 months of INH and RIF was cost saving compared with INH alone, and the cost savings increased as the prevalence of infection with strains resistant to INH increased (7). Instances of severe and fatal liver disease have been reported in patients taking RIF and PZA for treatment of latent tuberculosis infection (8). In addition, the frequency of hepatotoxicity has been shown to be greater with RIF--PZA than with INH alone (7.7% Grade 3 or 4 hepatotoxicity with RIF--PZA compared with 1% for INH; p = 0.001) (9). In view of these data, the regimen should be used with caution and with careful monitoring, measuring serum AST and bilirubin at baseline and after 2, 4, and 6 weeks of treatment. RIF--PZA is not recommended for patients with underlying liver disease or a history of alcoholism, or for those who have had hepatotoxicity from INH. The regimen should be reserved for patients who are not likely to complete a longer course of treatment and who can be monitored carefully. References
Untreated tuberculosis represents a far greater hazard to a pregnant woman and her fetus than does treatment of the disease. Infants born to women with untreated tuberculosis may be of lower birth weight than those born to women without tuberculosis and, rarely, the infant may acquire congenital tuberculosis (1--3). Thus, treatment of a pregnant woman with suspected tuberculosis should be started if the probability of tuberculosis is moderate to high. The initial treatment regimen should consist of INH, RIF, and EMB. SM should not be substituted for EMB. Although PZA is recommended for routine use in pregnant women by the WHO (4) and the IUATLD (5), the drug has not been recommended for general use in pregnant women in the United States because of insufficient data to determine safety. However, some public health jurisdictions in the United States have used PZA in pregnant women without reported adverse events (1). If PZA is not included in the initial treatment regimen, the minimum duration of therapy is 9 months. Pyridoxine, 25 mg/day, should be given to pregnant women who are receiving INH. INH, RIF, and EMB cross the placenta, but none has been shown to have teratogenic effects (6). SM, the only antituberculosis drug documented to have harmful effects on the human fetus, interferes with development of the ear and may cause congenital deafness. In 40 pregnancies among women being treated with SM, 17% of the babies had eighth nerve damage with deficits ranging from mild hearing loss to bilateral deafness (6,7). Kanamycin, amikacin, and capreomycin presumably share this toxic potential; however, there is little specific information on the fetal effects of these three drugs. PAS was used commonly with INH in the past and there was no indication of teratogenicity among babies whose mothers had received these two drugs (2). There are not enough data to determine the risk of cycloserine or ethionamide, although one report described nonspecific teratogenic effects attributed to ethionamide (8). The fluoroquinolones have been associated with arthropathies in young animals; therefore, they should be avoided if possible in pregnant women (6). In general, administration of antituberculosis drugs is not an indication for termination of pregnancy (2). However, in women who are being treated for drug-resistant tuberculosis, counseling concerning the risk to the fetus should be provided because of the known and unknown risks of the second-line agents. Breastfeeding should not be discouraged for women being treated with first-line agents, because the small concentrations of these drugs in breast milk do not produce toxic effects in the nursing infant (9). Conversely, drugs in breast milk should not be considered to serve as effective treatment for active tuberculosis or latent tuberculosis infection in a nursing infant. Supplementary pyridoxine is recommended for the nursing mother receiving INH. The administration of the fluoroquinolones during breastfeeding is not recommended, although, as of 1998, there have been no reported cases of adverse reactions in infants breast fed by women taking these drugs (6). References
8.7. Renal Insufficiency and End-stage Renal Disease Renal insufficiency complicates the management of tuberculosis because some antituberculosis medications are cleared by the kidneys. Management may be further complicated by the removal of some antituberculosis agents via hemodialysis. Thus, some alteration in dosing antituberculosis medications is commonly necessary in patients with renal insufficiency and end-stage renal disease (ESRD) receiving hemodialysis (Table 15). Decreasing the dose of selected antituberculosis drugs may not be the best method of treating tuberculosis because, although toxicity may be avoided, the peak serum concentrations may be too low. Therefore, instead of decreasing the dose of the antituberculosis agent, increasing the dosing interval is recommended (1). The general approach described in Table 15 involves either estimating or measuring creatinine clearance. Administration of drugs that are cleared by the kidneys to patients having a creatinine clearance of less than 30 ml/minute and those receiving hemodialysis are managed in the same manner, with an increase in dosing interval (C. Peloquin, personal communication). There are insufficient data to guide dosing recommendations for patients having a reduced creatinine clearance but not less than 30 ml/minute. In such patients standard doses should be used, but measurement of serum concentrations should be considered to avoid toxicity. RIF and INH are metabolized by the liver, so conventional dosing may be used in the setting of renal insufficiency (1--5). PZA is also metabolized by the liver but its metabolites (pyrazinoic acid and 5-hydroxy-pyrazinoic acid) may accumulate in patients with renal insufficiency (3,6). EMB is about 80% cleared by the kidneys and may accumulate in patients with renal insufficiency (7). A longer interval between doses with three times a week administration is recommended for PZA and EMB (3,7). INH, EMB, and PZA (as well as its metabolites) are cleared by hemodialysis to some degree, but only PZA and presumably its metabolites are dialyzed to a significant degree (3). RIF is not cleared by hemodialysis because of its high molecular weight, wide distribution into tissues, high degree of protein binding, and rapid hepatic metabolism (3). Therefore, supplemental dosing is not necessary for INH, RIF, or EMB. If PZA is given after hemodialysis, supplemental dosing is not required. In general, antituberculosis drugs should be given after hemodialysis to avoid any loss of the drugs during hemodialysis, and to facilitate DOT. Doses of streptomycin, kanamycin, amikacin, and capreomycin must be adjusted in patients with renal failure because the kidneys excrete essentially all of these drugs. Approximately 40% of the dose is removed with hemodialysis when these drugs are given just before hemodialysis (8). Far less drug is likely to be removed once the drugs have had time to distribute throughout the body, and some accumulation of the drugs should be anticipated. As with EMB and PZA, the dosing interval should be increased. In general, the dose should not be reduced because the drugs exhibit concentration-dependent bactericidal action (9), and smaller doses may reduce drug efficacy. Ethionamide is not cleared by the kidneys, nor is the drug removed with hemodialysis, so no dose adjustment is necessary (10). PAS is modestly cleared by hemodialysis (6.3%) but its metabolite, acetyl-PAS, is substantially removed by hemodialysis; twice daily dosing (4 g) should be adequate if the granule formulation is used (Jacobus Pharmaceuticals) (10). Cycloserine is excreted primarily by the kidney, and is cleared by hemodialysis (56%). Thus, an increase in the dosing interval is necessary to avoid accumulation between hemodialysis sessions, and the drug should be given after hemodialysis to avoid underdosing (10). The fluoroquinolones undergo some degree of renal clearance that varies from drug to drug. For example, levofloxacin undergoes greater renal clearance than moxifloxacin (11). It should be noted that the fluoroquinolone dosing recommendations for end-stage renal disease provided by the manufacturers were developed for treating pyogenic bacterial infections. These recommendations may not be applicable to the treatment of tuberculosis in patients with end-stage renal disease. As noted above, administration of all antituberculosis drugs immediately after hemodialysis will facilitate DOT (three times per week) and avoid premature removal of the drugs (2). It is important to monitor serum drug concentrations in persons with renal insufficiency who are taking cycloserine, EMB, or any of the injectable agents to minimize dose-related toxicity, while providing effective doses. Clinicians also should be aware that patients with end-stage renal disease may have additional clinical conditions, such as diabetes mellitus with gastroparesis, that may affect the absorption of the antituberculosis drugs, or they may be taking concurrent medications that interact with these drugs. Under these circumstances a careful clinical and pharmacologic assessment is necessary, and, in selected cases, serum drug concentration measurements may be used to assist in determining the optimum dose of the antituberculosis drugs (9). Finally, data currently do not exist for patients receiving peritoneal dialysis. Because the drug removal mechanisms differ between hemodialysis and peritoneal dialysis, it cannot be assumed that all of the recommendations in Table 15 will apply to peritoneal dialysis. Such patients may require close monitoring, including measurements of the serum concentrations of the antituberculosis drugs. References
The treatment of tuberculosis in patients with unstable or advanced liver disease is problematic for several reasons. First, the likelihood of drug-induced hepatitis may be greater. Second, the implications of drug-induced hepatitis for patients with marginal hepatic reserve are potentially serious, even life-threatening. Finally, fluctuations in the biochemical indicators of liver function (with/without symptoms) related to the preexisting liver disease confound monitoring for drug-induced hepatitis. Thus, clinicians may consider regimens with fewer potentially hepatotoxic agents in patients with advanced or unstable liver disease, and expert consultation is advisable in treating such patients. It should be noted that tuberculosis itself may involve the liver, causing abnormal liver function; thus, not all abnormalities in liver function tests noted at baseline should be attributed to causes other than tuberculosis. The hepatic abnormalities caused by tuberculosis will improve with effective treatment. Possible treatment regimens in the setting of liver disease include the following. 8.8.1. Treatment without INH As described in Section 5.2, Alternative Regimens, analysis of data from several studies conducted by the BMRC in patients with smear-positive pulmonary tuberculosis demonstrated high levels of efficacy with 6-month regimens despite in vitro resistance to INH so long as the initial phase contained four drugs and RIF was used throughout the 6 months (1). Subsequent studies by the Hong Kong Chest Service and the BMRC suggested that results were improved when PZA was used throughout the 6 months (2). Thus, it is reasonable to employ an initial phase regimen of RIF, PZA, and EMB followed by a continuation phase of RIF, EMB, and PZA (Rating BII). Although this regimen has two potentially hepatotoxic medications, it has the advantage of retaining the 6-month duration. 8.8.2. Treatment without PZA Although the frequency of PZA-induced hepatitis is slightly less than occurs with INH or RIF, the liver injury induced by this drug may be severe and prolonged (3). Therefore, one might elect to employ a regimen with an initial phase of INH, RIF, and EMB for 2 months followed by a continuation phase of INH and RIF for 7 months, for a total of 9 months (Table 2, Regimen 4). 8.8.3. Regimens with only one potentially hepatotoxic drug For patients with advanced liver disease, a regimen with only one potential hepatotoxic drug might be selected. Generally, RIF should be retained. Additional agents in such regimens could include EMB, a fluoroquinolone, cycloserine, and injectable agents. The duration of treatment with such regimens should be 12--18 months, depending on the extent of the disease and the response (Rating CIII). Consultation is advised in such situations. 8.8.4. Regimens with no potentially hepatotoxic drugs In the setting of severe unstable liver disease, a regimen with no hepatotoxic agents might be necessary. Such a regimen might include SM, EMB, a fluoroquinolone, and another second-line oral drug. There are no data that provide guidance as to the choice of agents or the duration of treatment or that indicate the effectiveness of such a regimen. Expert opinion suggests that a regimen of this sort should be given for 18--24 months (Rating CIII). Consultation should always be obtained before embarking on such a treatment plan. References
8.9. Other Associated Disorders Tuberculosis commonly occurs in association with other diseases or conditions. An associated disorder may alter immune responsiveness, thereby causing a predisposition to tuberculosis, or simply may be a disorder that occurs frequently in the same social and cultural milieu as tuberculosis. Examples of the former class of disorders include HIV infection, hematologic or reticuloendothelial malignancies, immunosuppressive therapy, chronic renal failure, poorly controlled, insulin-dependent diabetes mellitus, and malnutrition. Silicosis, by impairing pulmonary macrophage function, is a unique example of local immune dysfunction. The latter group of disorders includes chronic alcoholism and its secondary effects, other substance abuse, and psychiatric illnesses, among others. All of these conditions may influence the organization, supervision, and outcome of therapy (discussed in Section 2: Organization and Supervision of Treatment). The response of immunocompromised patients to treatment may not be as good as would be expected in a person with normal immunity, although in patients with HIV infection the response to treatment is not impaired Nevertheless, therapeutic decisions for the immunocompromised host should be more individualized, taking into account the severity of tuberculosis and the response to treatment. When possible, steps should be taken to correct the immune deficiency. In patients with silicotuberculosis there are data demonstrating that the rate of cure is improved if the continuation phase is extended for at least 2 months (1,2). References
9. Management of Relapse, Treatment Failure, and Drug Resistance9.1. Relapse Relapse refers to the circumstance in which a patient becomes and remains culture-negative while receiving antituberculosis drugs but, at some point after completion of therapy, either becomes culture-positive again or experiences clinical or radiographic deterioration consistent with active tuberculosis. In such patients vigorous efforts should be made to establish a diagnosis and to obtain microbiological confirmation of the relapse to enable testing for drug resistance. True relapses are due to failure of chemotherapy to sterilize the host tissues, thereby enabling endogenous recrudescence of the original infection. In some hyperendemic settings, however, exogenous reinfection with a new strain of M. tuberculosis may be responsible for the apparent relapse (1). Patients who are most likely to have true relapses are those with extensive tuberculosis whose sputum cultures remain positive after 2 months of chemotherapy (2--4). Most patients relapse within the first 6--12 months after completion of therapy. In nearly all patients with tuberculosis caused by drug-susceptible organisms who were treated with rifamycin-containing regimens using DOT, relapses occur with susceptible organisms (5,6). However, in patients who received self-administered therapy or a nonrifamycin regimen and who have a relapse, the risk of acquired drug resistance is substantial. In addition, if initial drug susceptibility testing was not performed and the patient fails or relapses with a rifamycin-containing regimen given by DOT, there is a high likelihood that the organisms were resistant from the outset. Among patients who received self-administered therapy, the risk of erratic drug administration leading to relapse with resistant organisms is greater. In view of these considerations, the selection of empirical treatment regimens for patients with relapses should be based on the prior treatment scheme. For patients with tuberculosis that was caused by drug-susceptible organisms, who were treated by DOT, and who have relapses, retreatment using the standard four-drug initial phase regimen may be appropriate, at least until the results of susceptibility tests are known. For patients who did not receive DOT or are known to have had irregular treatment in the past, it is prudent to infer a higher risk of acquired drug resistance and begin an expanded regimen (see below). The expanded regimen is indicated especially in patients with impaired immunity, limited respiratory reserve, central nervous system involvement, or other life-threatening circumstances, that is, cases in which treatment with an inadequate regimen could have severe consequences. For the relatively few patients in whom epidemiologic circumstances provide a strong suspicion of exogenous reinfection as the cause of apparent relapse, the choice of a regimen is influenced by the drug susceptibility pattern of the presumed source case. If the presumed source case is known to have tuberculosis caused by drug-susceptible organisms, resumption of a standard four-drug initial phase may be indicated. However, if the likely source case is known to have drug-resistant organisms, an empirically expanded regimen based on the resistance profile of the putative source case may be suitable. There are no clinical trials to guide the choice of agents to include in expanded empirical regimens for presumed drug resistance; however, expert opinion indicates that such regimens should generally employ INH, RIF, and PZA plus an additional three agents, based on the probability of in vitro susceptibility. Usual agents would include EMB, a fluoroquinolone, and an injectable agent such as SM (if not used previously, and the initial isolate was susceptible) amikacin, kanamycin or capreomycin, with or without other drugs. 9.2. Treatment Failure Treatment failure is defined as continued or recurrently positive cultures in a patient receiving appropriate chemotherapy. Among patients with drug-susceptible pulmonary tuberculosis, even with extensive lung cavitation, 90--95% will be culture-negative after 3 months of treatment with a regimen that contains INH and RIF. During this time the vast majority of patients show clinical improvement, including defervescence, reduced cough, and weight gain. Thus, patients with persistently positive cultures after 3 months of chemotherapy, with or without on-going symptoms, should be evaluated carefully to attempt to identify the cause of the delayed response. Patients whose sputum cultures remain positive after 4 months of treatment are considered to have failed treatment. There are multiple potential reasons for treatment failure. If the patient is not receiving DOT, the most likely explanation for persistently positive cultures is nonadherence to the drug regimen. Among patients receiving DOT, cryptic nonadherence (spitting out or deliberately regurgitating pills) or failure of the health care system to reliably deliver the drugs may be the cause. Other potential reasons include unrecognized drug resistance (Was initial drug-susceptibility testing done? Was it reported accurately?), malabsorption (prior resectional surgery of the stomach or small intestine, taking tuberculosis medication with antacids or other drugs/substances that might bind or interfere with drug absorption (see Section 6.1: Drug Administration, and Section 7.1: Interactions Affecting Antituberculosis Drugs), or simply an extreme biologic variation (For unclear reasons, rare "normal" patients may experience very protracted disease including persistently positive cultures or prolonged symptoms in the face of chemotherapy that would be expected to be effective). Laboratory error should also be considered as a possible reason for a positive culture in a patient who is doing well. Recent reports document cross contamination or mislabeling of specimens as a source for some of these unexpectedly positive cultures (7,8). Clinicians should be alert, as well, to the possibility of transient clinical or radiographic worsening (paradoxical reactions), despite appropriate therapy that would eventually result in cure. Examples of this include ongoing inflammation at sites of lymphadenitis, worsened abnormalities on chest radiographs after several months of treatment, or the new appearance of pleural effusions during therapy for pulmonary tuberculosis (9--11). Such paradoxical worsening during treatment occurs more commonly but not exclusively in persons with HIV infection (12--14) (see Section 8.1: HIV Infection). For patients who meet criteria for treatment failure, the possible reasons listed above should be addressed promptly. If clinicians are not familiar with the management of drug-resistant tuberculosis, prompt referral to, or consultation with a specialty center is indicated. If treatment failure is presumed to be due to drug resistance and the patient does not have severe tuberculosis, one may either initiate an empirical retreatment regimen or wait for drug susceptibility results from a recent isolate. If the patient is seriously ill or has a positive sputum AFB smear, an empirical regimen that would be anticipated to be effective should be started immediately and continued until susceptibility tests are available to guide therapy. For patients who have failed treatment, mycobacterial isolates should be sent promptly to a reference laboratory for susceptibility testing for both first- and second-line drugs. A fundamental principle in managing patients who have failed treatment is that a single new drug should never be added to a failing regimen; so doing may lead to acquired resistance to the added drug. In such cases, it is generally prudent to add at least three new drugs to which susceptibility could logically be inferred to lessen the probability of further acquired resistance. As noted previously there are no clinical trials to guide the choice of an empirical regimen; however, expert opinion indicates that empirical retreatment regimens might include a fluoroquinolone such as levofloxacin, an injectable agent such as SM (if not used previously and the isolate was susceptible initially), amikacin, kanamycin, or capreomycin, and an oral agent such as PAS, cycloserine, or ethionamide (Rating AIII). When drug susceptibility results are available, the regimen should be adjusted according to the results. 9.3. Management of Tuberculosis Caused by Drug-Resistant Organisms Tubercle bacilli are continually undergoing spontaneous mutations that create resistance to individual antituberculosis drugs. However, the frequency of these single mutations is sufficiently low that with appropriate combination chemotherapy that is reliably ingested, clinically significant resistance will not develop (see Section 4.1: Combination Chemotherapy) (15). Most commonly the development of acquired drug resistance occurs when there is a large bacillary population, such as in pulmonary cavities, when an inadequate drug regimen is prescribed (inappropriate drugs, insufficient dosage) or when there is a combined failure of both the patient and the provider to ensure that an adequate regimen is taken (16). Rarely, malabsorption of one or more antituberculosis drugs may account for acquired resistance. Drug resistance is much more likely to occur in cavitary pulmonary tuberculosis because of the immense number of rapidly multiplying bacilli in the cavity(ies) (17). During extended or repeated treatment, resistance to multiple agents may evolve. Patients with acquired drug resistance may transmit their strains to others who, if they develop tuberculosis, will have primary drug resistance. Drug resistance in a patient with newly diagnosed tuberculosis may be suspected on the basis of historical (previous treatment) or epidemiologic information (contact with a known drug-resistant case or coming from a region in which drug resistance is common) (18,19). In such situations it is prudent to employ an empirically expanded regimen, as described previously, especially if the patient is seriously ill (Table 16). Drug resistance can be proven only by drug-susceptibility testing performed in a competent laboratory (Table17). The steps taken when resistance is shown to be present are of critical importance. Patients harboring strains of M. tuberculosis resistant to both INH and RIF (MDR) are at high risk for treatment failure and further acquired resistance; they must be referred immediately to a specialist or consultation obtained from specialized treatment centers. Patients with strains resistant to RIF alone have a better prognosis than MDR cases, but also are at increased risk for failure and additional resistance. Thus, their management should also be subject to special scrutiny. Definitive randomized or controlled studies have not been performed among patients with the various patterns of drug resistance. In the absence of ideal evidence, practices in the treatment of patients are based on a mixture of general principles, extrapolations and expert opinion. The WHO and IUATLD have formulated standard algorithmic regimens for the management of treatment failure or chronic cases, largely based on the principles listed below, as well as on expert opinion (20,21). This approach is best suited to regions without in vitro susceptibility testing capacity and access to the full array of retreatment medications, but it is not appropriate for industrialized nations with more ample resources (22,23). Guidelines for management of patients with tuberculosis caused by drug-resistant organisms are based on the following guidelines, all of which are rated A III:
Table 16 contains regimens suggested for use in patients with various patterns of drug-resistant tuberculosis. 9.4. Role of Surgery in MDR Tuberculosis The role of resectional surgery in the management of patients with extensive pulmonary MDR tuberculosis has not been established in randomized studies. In one series, patients with severe drug resistance (on average, having resistance to more than 5 drugs) appeared to benefit from the resection of cavitary or badly damaged lung tissue when compared with historical controls (31). In contrast, other clinicians have reported patients with drug resistance having similar cure rates without surgery (25,32). The disparity in these reports may be due to long-standing disease with extensive fibrosis in the former group. If surgery is to be done, it should be performed by an experienced surgeon after the patient has received several months of intensive chemotherapy. Even with successful resection, 12--24 additional months of chemotherapy, using drugs to which there is demonstrated susceptibility, should be given. References
   9.5 Laboratory Considerations in Determining Drug Resistance Susceptibility testing of M. tuberculosis is critical for appropriate patient management and should be performed on an initial isolate from all patients from whom M. tuberculosis is recovered (1). Public health laboratories routinely will perform susceptibility testing on initial isolates but, often, private laboratories do not perform such testing unless specifically requested to do so by the physician. As noted previously, susceptibility testing should be repeated if the patient still has a positive culture result after 3 months of therapy or again develops positive cultures after a period of negative cultures (2). Antimicrobial susceptibility testing should be performed using a standard methodology, such as that recommended by the National Committee for Clinical Laboratory Standards (3). The second edition of a tentative standard (M24-T2) for susceptibility testing of mycobacteria was published by the National Committee for Clinical Laboratory Standards in 2000 (3). Susceptibility of M. tuberculosis is determined by evaluating the ability of an isolate to grow on agar or in broth containing a single "critical" concentration of a drug (2). The agar proportion method has been proposed as the reference method for all antituberculosis drugs except pyrazinamide, in which case the BACTEC broth-based methodology is the reference method (3). With the agar proportion method, resistance is defined as growth on the drug-containing plate that is more than 1% of the growth on the non--drug-containing plate (4). Because the agar method requires up to 6 weeks to yield results, it is recommended that initial susceptibility testing of M. tuberculosis isolates to first-line antituberculosis drugs be performed using more rapid broth-based methods (e.g., BACTEC and others). The goal, as stated by CDC, is to have culture and susceptibility results (to first-line drugs) available within 28 days of receipt of a clinical specimen (5). The critical concentrations recommended by the National Committee for Clinical Laboratory Standards for agar proportion method and "equivalent" concentrations for broth-based testing methods are shown in Table 17 (2,3). The National Committee for Clinical Laboratory Standards recommends that susceptibility testing be performed for INH (two concentrations) and RIF and EMB (one concentration each) using a broth-based method on all initial M. tuberculosis isolates. Pyrazinamide testing may be done if there is a sufficiently high prevalence of PZA resistance. It is also recommended that the full panel of drugs (including second-line drugs) be tested when there is resistance to RIF alone or to two or more drugs. Testing of second-line drugs is performed using the agar proportion method, generally by public health laboratories. Secondary antituberculous drugs used for testing are capreomycin, ethionamide, kanamycin (which also predicts amikacin susceptibility), ofloxacin (used to assess fluoroquinolone activity), PAS, rifabutin, and SM (3). For second-line drug testing, a second concentration of EMB is also recommended. Susceptibility testing for cycloserine is not recommended because of the technical problems associated with the test. References
 10. Treatment Of Tuberculosis in Low-Income Countries: Recommendations and Guidelines of the WHO and the IUATLDThis brief summary of the differences between the recommendations for treatment of tuberculosis in high-income, low-incidence countries and low-income, high incidence countries is presented to provide an international context for the ATS/CDC/IDSA guidelines. As tuberculosis in low-incidence countries, such as the United States, becomes more and more a reflection of the situation in high-incidence countries, it is important that health care providers in low-incidence countries have an understanding of the differences in the approaches used and the reasons for these differences so as to be better equipped to treat the increasing proportion of patients from high-incidence countries (1). As noted at the outset of this document, the ATS/CDC/IDSA recommendations cannot be assumed to be applicable under all epidemiologic and economic circumstances. The incidence of tuberculosis and the resources with which to confront it to an important extent determine the approaches used. A number of differences exist between these new ATS/CDC/IDSA recommendations, and the current tuberculosis treatment recommendations of WHO (2) and IUATLD (3), the two major sets of international guidelines. Rather than being recommendations per se, the IUATLD document presents a distillation of IUATLD practice, validated in the field. The WHO and the IUATLD documents target, in general, countries in which mycobacterial culture and susceptibility testing and radiographic examinations are not widely available. These organizations recommend a tuberculosis control strategy called "DOTS" (Directly Observed Treatment, Short-Course) in which direct observation of therapy ("DOT" in the current statement) is only one of five key elements (4). The boxed insert lists the elements of DOTS strategy. Selected important differences among the recommendations are summarized below. Some of the differences arise from variations in strategies, based on availability of resources, whereas others, such as the use of twice weekly regimens, arise from different interpretations of common elements, for example, whether DOT is used throughout the entire course of therapy or is limited to the initial phase. 10.1. Microbiological Tests for Diagnosis and Evaluation of Response The WHO and the IUATLD recommend diagnosis and classification of cases and assessment of response based on sputum AFB smears. The AFB smear is emphasized because access to reliable culture facilities is limited in many countries. In addition, the AFB smear identifies patients who are most likely to transmit the organism. Susceptibility testing for new patients is not recommended because of cost, limited applicability and lack of facilities. However, susceptibility testing is recommended by the WHO for patients who fail (sputum smear--positive in month 5 of treatment or later during the course of treatment) the initial treatment regimen, and for those who fail a supervised retreatment regimen. Regarding follow-up, it is recommended by the WHO and the IUATLD that patients who have initial positive smears have repeat smears examined at 2 months, 5 months, and at completion of treatment (either 6 or 8 months). The IUATLD recommends that for patients who have positive smears at 2 months, the initial phase should be extended for 1 month. 10.2. Use of Chest Radiographs in Diagnosis and Follow-Up of Patients Being Treated In many parts of the world radiographs are not readily available. Moreover, because the highest priority for treatment is the highly infectious sputum smear--positive patient, there is concern that treatment based on radiographic findings alone is an inefficient use of resources. Thus, chest radiography is recommended by both the WHO and the IUATLD only for patients with negative sputum smears and is not recommended at all for follow-up. 10.3. Initial Treatment Regimens The WHO recommends a single initial phase of daily INH, RIF, PZA, and EMB (or SM) for 2 months followed by a continuation phase of either daily or three times a week INH and RIF, all given by DOT, for 4 months or daily INH and EMB for 6 months (self-administered). The WHO specifically discourages programs from using twice weekly regimens, the reason being that there is a lesser margin of safety if a dose or doses are missed. The IUATLD recommends a 2-month initial phase of INH, RIF, PZA, and EMB given by DOT, followed by a 6-month continuation phase of daily INH and thiacetazone, self-administered. For patients with HIV infection the IUATLD recommends EMB in place of thiaocetazone. The IUATLD also recommends a 12-month regimen with a 2-month initial phase of INH, SM, and thioacetazone given daily and a 10-month continuation phase of daily INH and thioacetazone. This regimen is intended to be used for patients who have negative smears or when the 8-month regimen is not available. The rationale for the 8-month regimen recommendation is that it is felt that RIF should always be given by DOT; yet, many programs cannot afford to provide the supervision required by DOT for the full 6 months of treatment. The 8-month regimen is less efficacious in patients with drug-susceptible tuberculosis, but use of this regimen will likely preserve RIF for use in retreatment regimens. In addition to the issue of supervision, the 8-month regimen's continuation phase of INH and EMB costs about 27% less than a 4-month continuation phase of daily INH and RIF. 10.4. Approach to Previously Treated Patients The WHO and the IUATLD recommend a standardized regimen for patients who have relapsed, had interrupted treatment, or have failed treatment. (The approach to this last group of patients is currently under discussion at the WHO.) The regimen consists of an initial phase of INH, RIF, PZA, EMB, and SM given daily for 2 months and then 1 month of daily INH, RIF, PZA, and EMB. The continuation phase consists of 5 months of daily INH, RIF, and EMB. Patients who have failed supervised retreatment are considered "chronic" cases and are highly likely to have tuberculosis caused by MDR organisms. Susceptibility testing and a tailored regimen using second-line drugs based on the test results are recommended by the WHO, if testing and second-line drugs are available (5). The IUATLD recommendations do not address the issue. The issue of chronic cases is an area of considerable controversy (6). In countries with sufficient resources, such as the United States, individualized retreatment regimens, based on drug susceptibility patterns, as described in Section 9, Management of Relapse, Treatment Failure, and Drug Resistance, are recommended. However, in countries without the capacity to obtain susceptibility tests, individualized regimens cannot be prescribed. Nevertheless, at least one group has demonstrated that in a high-incidence, low-income country (Peru) treatment with individualized regimens is feasible and effective (7). 10.5. Monitoring of Outcomes of Therapy Both the WHO and the IUATLD recommend a formal system for monitoring outcomes of treatment that classifies all cases into one of six categories (cured, completed without proof of cure, failed, died, defaulted, or transferred out). The assessment of cure is based on clinical response and on sputum AFB smear (or culture when available) at completion of treatment. The analysis of these outcomes is by temporal cohorts and enables identification of programmatic shortcomings. 10.6. Recommended Doses of Antituberculosis Drugs The WHO recommends 10 mg/kg as the dose for three times weekly INH, whereas the ATS/CDC/IDSA recommend 15 mg/kg (Table 3). There is no difference in the daily doses recommended for adults (5 mg/kg per day to a maximum of 300 mg/day), but the ATS/CDC/IDSA recommend a higher dose for children (10--15 mg/kg per day), based primarily on the expert opinion of pediatricians. The IUATLD recommendations are based on the number of pills required for three weight ranges resulting in a dose of about 5 mg/kg up to 300 mg/day. The clinical trials of the BMRC that established the efficacy of three times weekly regimens all used an INH dose of 15 mg/kg. The 10-mg/kg INH dose for thrice-weekly regimens was extrapolated by the WHO and the IUATLD (with assistance from global experts), and was chosen to maintain the weekly amount of INH approximately equal to that of the daily or twice weekly regimens. 10.7. Drugs/Preparations Not Available in the United States Thioacetazone, which formerly was commonly used, is still available in most parts of the world, but is used less frequently. However, thioacetazone remains listed as an "essential" first-line drug by the WHO and is a component of the recommended IUATLD first-line regimen. Combination preparations not available in the United States but listed by the WHO include the following: INH (150 mg) and EMB (400 mg); INH (100 mg) and thioacetazone (50 mg); and INH (75 mg), RIF (150 mg), PZA (400 mg), and EMB (275 mg). The IUATLD recommends using only combination preparations of INH and RIF or INH and thiacetazone. 10.8. Treating Pregnant Women Both the WHO and the IUATLD include PZA in the regimen for treating pregnant women, in the absence of data indicating that there are adverse consequences. 10.9. Management of Common Adverse Reactions Neither baseline nor follow-up testing is recommended by the WHO and the IUATLD. It is recommended that patients be taught to recognize the symptoms associated with drug toxicity and to report them promptly. References
 11. Research Agenda for Tuberculosis Treatment11.1. New Antituberculosis Drugs New antituberculosis drugs are needed for three reasons: to shorten or otherwise simplify treatment of tuberculosis caused by drug-susceptible organisms, to improve the treatment of patients with MDR tuberculosis, and to provide more effective and efficient treatment of latent tuberculosis infection (LTBI) (1). Although treatment regimens for drug-susceptible tuberculosis are effective, they must be administered for a minimum of 6 months to achieve optimal results. Nonadherence to this relatively lengthy course of treatment remains a major problem. To address the problem of nonadherence, DOT (as a component of the DOTS strategy) is recommended as a standard of care worldwide. However, the administrative and financial burden of providing DOT for all patients is considerable. Thus, new drugs that would permit significant shortening of treatment are urgently needed, as are drugs that could enable effective treatment to be given at dosing intervals of 1 week or more. Rates of multidrug-resistant tuberculosis are alarmingly high in several countries (2), and even in countries, such as the United States, where the rates are low and decreasing, the occasional case presents an often extremely difficult treatment problem (see Section 9: Management of Relapse, Treatment Failure, and Drug Resistance). Current treatment regimens for drug-resistant tuberculosis utilize drugs that are less effective, more toxic, and more expensive than those used for standard treatment. Moreover, these treatment regimens often have to be given for 18--24 months. Although new drugs that are effective against resistant organisms would alone not solve the problem of drug resistance, their judicious use would greatly improve the treatment for many patients. Finally, the United States and several other low-incidence countries have embarked on plans to eliminate tuberculosis. An important component of an elimination strategy is the identification and treatment of persons with LTBI who are at high risk of developing tuberculosis (3). In the United States the most commonly used LTBI treatment regimen is INH given for 9 months; however, poor adherence to this regimen imposes a major limitation on its effectiveness. A shorter LTBI treatment regimen with RIF and PZA appears to be effective, but reports have indicated that toxicity may be unacceptably high (4). Thus, new drugs to provide for safe and effective "short-course" LTBI treatment are a major need. No truly novel compounds that are likely to have a significant impact on tuberculosis treatment are presently available for clinical study. However, further work to optimize the effectiveness of once weekly rifapentine regimens and investigate the role of newer fluoroquinolones in the treatment of drug-susceptible tuberculosis is warranted. As noted above, once weekly rifapentine--INH is recommended only in the continuation phase for HIV-negative patients with noncavitary pulmonary tuberculosis who have negative sputum smears at completion of 2 months of treatment. Two approaches to improve intermittent rifapentine regimens have been suggested by experimental studies: increasing the rifapentine dosage (5), and adding moxifloxacin as a companion drug to provide better protection against the development of drug resistance and enhance the sterilizing activity of the regimen (6). Other data from a clinical trial of ofloxacin suggest that fluoroquinolones have the potential to significantly shorten treatment (7). Of the newer fluoroquinolones with more potent activity against M. tuberculosis, moxifloxacin appears to be the most promising. Other compounds that might become available for clinical evaluation in the future include the nitroimidazopyrans that are chemically related to metronidazole, for which activity against dormant M. tuberculosis has been suggested; oxazolidinones such as linezolid; and drugs that target isocitrate lyase, an enzyme that may be necessary for the establishment of latent tuberculosis infection (8). The nitroimidazopyran compound PA-824 has bactericidal activity comparable to that of INH and appears to act as well on bacilli maintained in an anaerobic environment (9). However, additional preclinical evaluation of PA-824 is needed before clinical studies could begin. Although linezolid, a drug that is marketed for the treatment of selected acute bacterial infections, does have demonstrated activity against M. tuberculosis, other compounds in that class may be more suited for the treatment of tuberculosis (10). 11.2. Other Interventions To Improve the Efficacy of Treatment A number of other approaches have been suggested that might lead to improved treatment outcome, including alternative drug delivery systems and a variety of methods of immunomodulation and immunotherapy. Experimental studies have demonstrated that effective serum concentrations of INH and PZA can be provided through incorporation of drug into slow-release, biodegradable polymers that are implanted subcutaneously (11). However, there has been little apparent commercial interest in pursuing this approach. Liposomal encapsulation of antituberculosis drugs has been suggested as an approach to direct drug to the proposed site of infection (i.e., the macrophage) providing for more effective and better tolerated therapy, as well as for more widely spaced treatment. Similarly, incorporation of drug into inhalable microparticles may reduce dose requirements, minimize toxicity, and deliver drug to infected alveolar macrophages. Although experimental studies have suggested that these approaches might be effective, little clinical work has been done in these areas (11,12). Because of possible detrimental effects of the cytokine, tumor necrosis factor-a, in HIV-associated tuberculosis, there has been some interest in the use of drugs, such as thalidomide and pentoxifylline, that block tumor necrosis factor-a production. Studies have shown that administration of thalidomide improves weight gain in both HIV-positive and HIV-negative tuberculosis patients (13). Pentoxifylline has been associated with reductions in circulating HIV viral load in patients with tuberculosis (14). However, the potential side effects of these drugs may outweigh possible benefits. A more promising intervention is the administration of "protective" cytokines, such as aerosolized interferon-g and subcutaneous interleukin-2, that have shown activity as adjuncts to chemotherapy in patients with multidrug-resistant tuberculosis (15,16). Another method of immunomodulation, the use of heat-killed preparations of M. vaccae as a therapeutic vaccine, has not shown clinically significant benefits when carefully evaluated in randomized clinical trials (17). Nonetheless, there continues to be interest in this approach, especially for patients with advanced drug-resistant tuberculosis. Other vaccines that have been shown to lead to expression of protective cytokines have shown more promise in experimental studies (18). Finally, a study suggested that the administration of Vitamin A and zinc to patients with pulmonary tuberculosis is associated with an increased rate of sputum conversion and improvement in chest radiographs (19). Further assessment of nutritional supplements in tuberculosis treatment may be indicated. 11.2.1. Better methods to identify and manage high- and low-risk patients As noted above, sputum culture positivity at 2 months appears to be a marker for an increased risk of relapse for patients with pulmonary tuberculosis. Surrogate markers that could be measured earlier in therapy and have a greater sensitivity and specificity for a poor outcome could better select high risk patients for more intensive or longer therapy, thus minimizing the likelihood of relapse. Studies of several molecular markers in the sputum have shown promise and deserve further evaluation (20). Conversely, markers that reliably identify patients at lower risk of an adverse treatment outcome would be helpful to select patients for less intense or shorter treatment. Whether or not low-risk patients can be treated with shorter regimens using currently available drugs is a topic of considerable importance. 11.2.2. Health services research to facilitate treatment administration and improve treatment outcome Although DOT (as a component of DOTS) is widely advocated as a universal standard of care for tuberculosis treatment, many tuberculosis control programs do not have the resources to provide DOT for all patients. Moreover, some programs have achieved excellent results by targeting DOT to patients known or suspected of being at increased risk for nonadherence. Further evaluation of alternatives to universal DOT is needed. Finally, although limited work has been done in the area of behavioral studies of tuberculosis patients and providers, an ambitious research agenda established in the mid-1990s has not been implemented and should be revisited (21). References
Acknowledgment The Committee thanks Elisha Malanga of the American Thoracic Society for excellent administrative support. The Committee also thanks the members of an ad hoc review panel convened by the Division of Tuberculosis Elimination, National Center for HIV, STD, and TB Prevention, CDC, for their thorough review and helpful comments. The members of this panel were as follows: Naomi Bock, M.D., James Burns, M.D., Mike Holcombe, M.P.P.A., James Lamberti, M.D., Evelyn Lancaster, R.N., Kathleen Moser, M.D., M.P.H., James McDaniel, M.D., F.A.C.P., Sonal Munsiff, M.D., Margaret Oxtoby, M.D., Carol Pozsik, R.N., M.P.H., Susan Ray, M.D., Jon Tillinghast, M.D., M.P.H., Victor Tomlinson, Jr., M.D., and Charles Wallace, Ph.D.
Joint Committee of the American Thoracic Society (ATS), the Infectious Diseases Society
of America (IDSA), and CDC Co-chairs: Henry M. Blumberg, M.D., IDSA; Philip C. Hopewell, M.D., ATS; Richard J. O'Brien, M.D., CDC. Members: William J. Burman, M.D.; Richard E. Chaisson, M.D.; Charles L. Daley, M.D.; Wafaa El Sadr, M.D., M.P.H.; Sue C. Etkind, B.S.N., M.S.; Lloyd N. Friedman, M.D.; Paula Fujiwara, M.D., M.P.H.; Malgosia Grzemska, M.D.; Michael D. Iseman, M.D.; Robert M. Jasmer, M.D.; Venkatarama R. Koppaka, M.D., Ph.D.; Richard I. Menzies, M.D.; Randall R. Reves, M.D.; Lee B. Reichman, M.D., M.P.H.; Patricia M. Simone, M.D.; Jeffrey R. Starke, M.D.; Andrew A. Vernon, M.D., M.H.S. * Charles Peloquin, Pharm.D., also made substantial contributions to the document. Table 1 Return to top. Figure 1 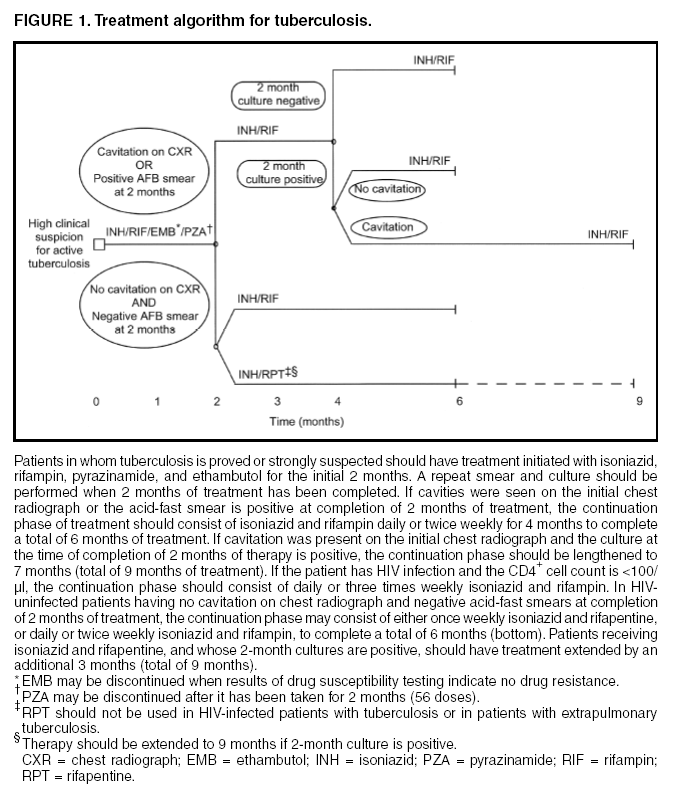 Return to top. Table 2 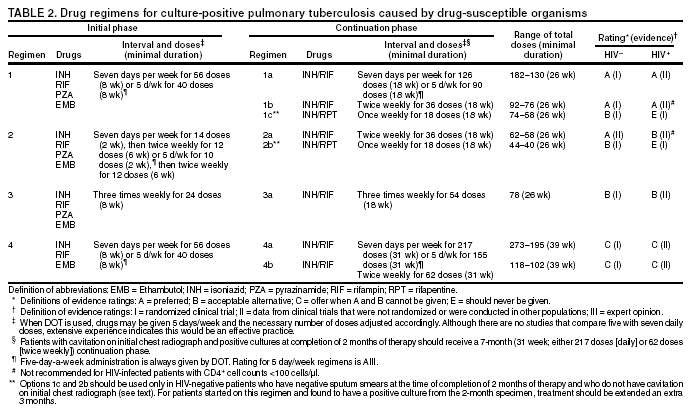 Return to top. Figure 2  Return to top. Table 3 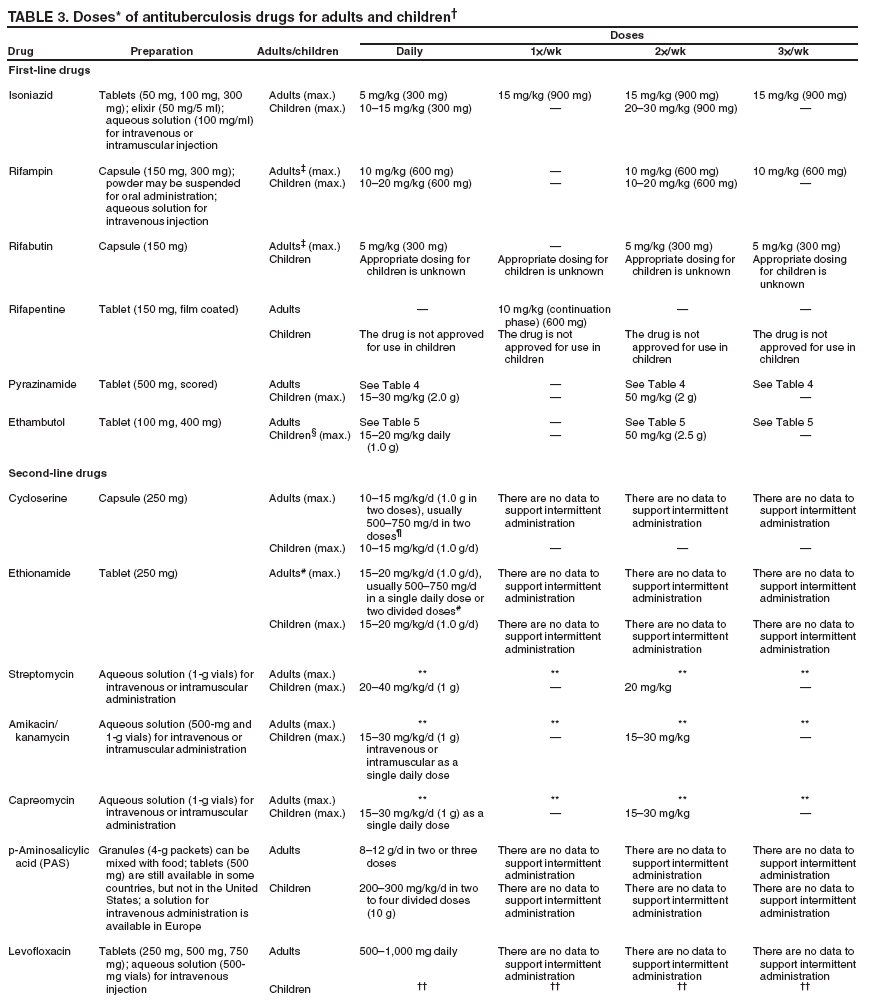 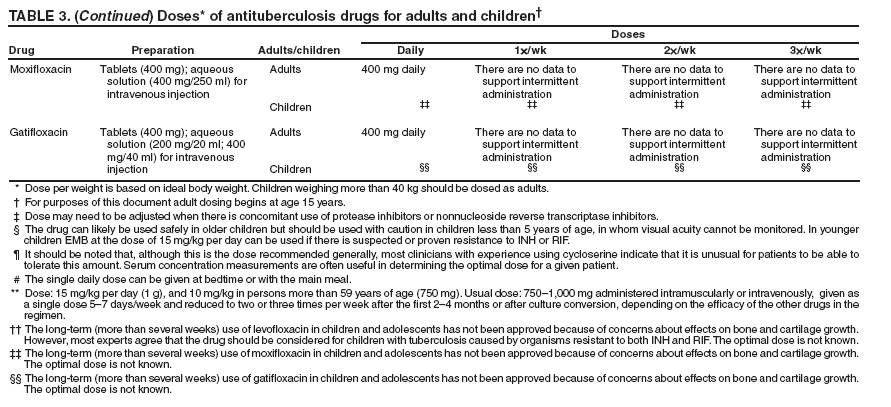 Return to top. Figure 3 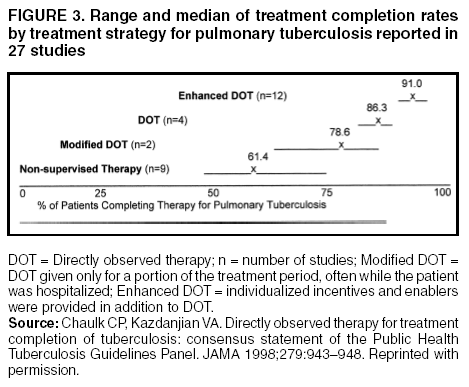 Return to top. Table 4 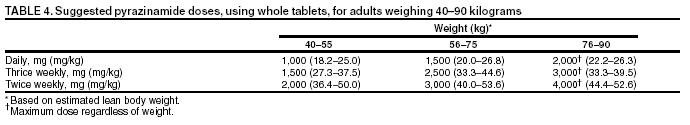 Return to top. Figure 4  Return to top. Table 5 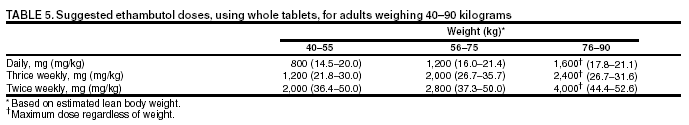 Return to top. Figure 5 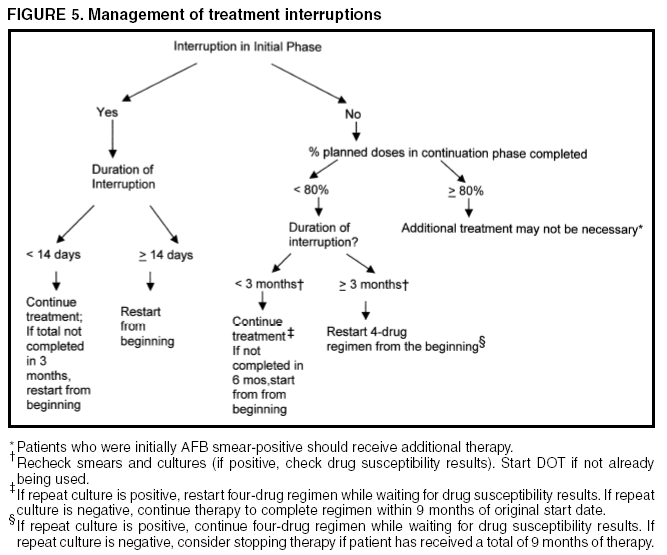 Return to top. Table 6  Return to top. Table 7  Return to top. Table 8  Return to top. Table 9  Return to top. Table 10  Return to top. Table 11  Return to top. Table 12 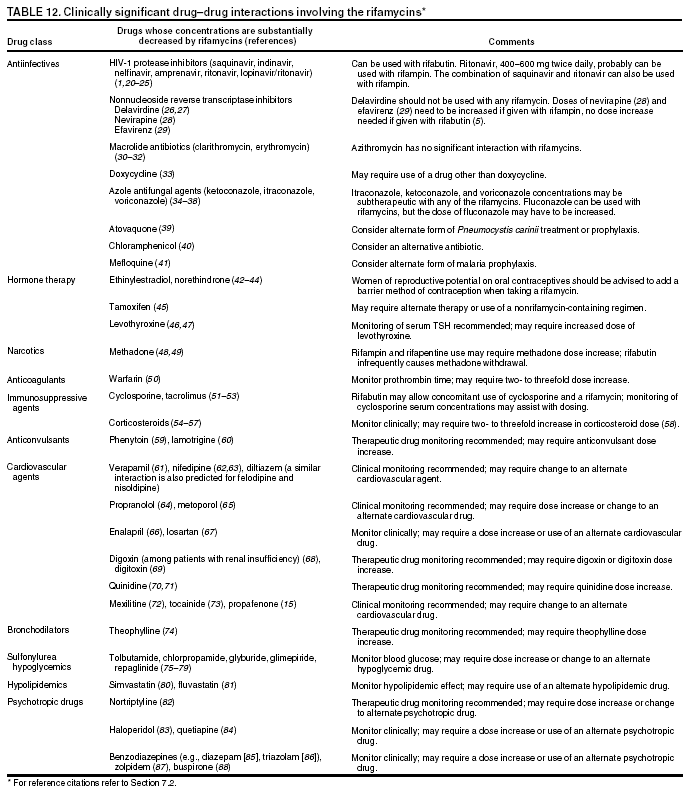 Return to top. Table 13 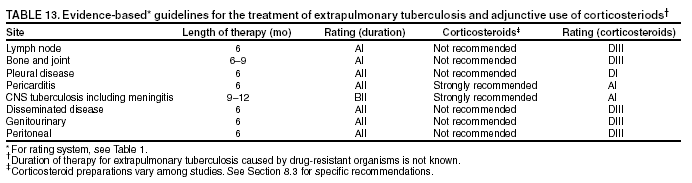 Return to top. Table 14  Return to top. Table 15 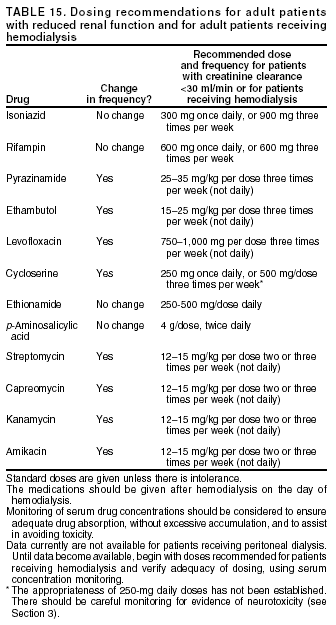 Return to top. Table 16  Return to top. Table 17 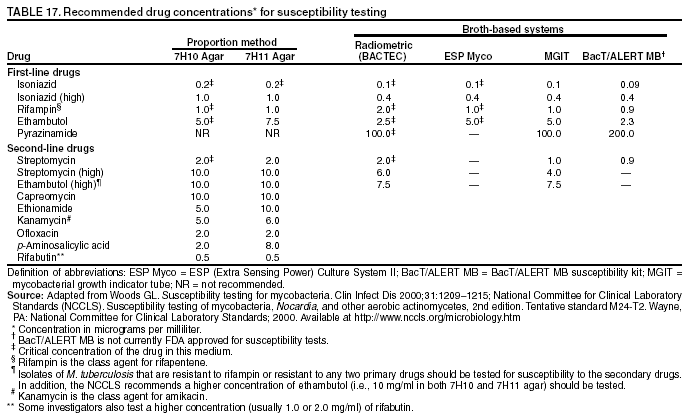 Return to top.
All MMWR HTML versions of articles are electronic conversions from ASCII text into HTML. This conversion may have resulted in character translation or format errors in the HTML version. Users should not rely on this HTML document, but are referred to the electronic PDF version and/or the original MMWR paper copy for the official text, figures, and tables. An original paper copy of this issue can be obtained from the Superintendent of Documents, U.S. Government Printing Office (GPO), Washington, DC 20402-9371; telephone: (202) 512-1800. Contact GPO for current prices. **Questions or messages regarding errors in formatting should be addressed to mmwrq@cdc.gov.Page converted: 6/4/2003 |
|||||||||
This page last reviewed 6/4/2003
|