 |
|
|
|
|
|
|
| ||||||||||
|
|
|
|
|
|
|
||||
| ||||||||||
|
|
|
|
|
Persons using assistive technology might not be able to fully access information in this file. For assistance, please send e-mail to: mmwrq@cdc.gov. Type 508 Accommodation and the title of the report in the subject line of e-mail. Nosocomial Bacteremias Associated with Intravenous Fluid Therapy -- USAMMWR 1971;20 (Special Supplement) (March 6, 1971) Between October 1970 and March 1, 1971, eight United States Hospitals in seven states experienced 150 bacteremias caused by Enterobacter cloacae or Gram-negative organisms of the Erwinia group. There were nine deaths; all were associated with intravenous (IV) fluid therapy. The Enterobacter bacteremias in all hospitals were substantially increased as compared to previous time periods. Four hospitals which isolated and identified Erwinia had not previously encountered infections with these organisms. In-depth epidemiologic investigations were performed in three of the hospitals (Figure_1). All eight hospitals utilize fluids and systems manufactured by Abbott Laboratories, which produces approximately 45 percent of all IV fluids sold within the United States. In approximately 30 cases, the same organisms were isolated from blood cultures and contaminated in-use IV fluids. In hospital B (see arrow, Figure_1) no further cases were observed after discontinuance of Abbott products. Enterobacter cloacae is occasionally encountered as an agent of bacteremia in American hospitals. However, unless fully speciated, this organism will not be identified. Erwinia, most well known as a plant pathogen, has only very rarely been isolated from human infection (1). Erwinia may be confused with members of the Klebsiella-Enterobacter group, and a rather detailed series of biochemical tests, with special emphasis on decarboxylase reactions, are needed to reliably differentiate the organisms. These septicemias were constantly characterized by intermittent high fever, although shock was infrequent. Young individuals or other patients without predisposing host factors were frequently afflicted. The great majority of cases simultaneously manifested extensive phlebitis at the site of infusion which occurred even when polyethylene catheters had been in place for only brief periods, and also occurred where only scalp vein needles were used. Discontinuance of IV therapy has resulted in dramatic clinical improvement; if such therapy is continued, however, antimicrobials have frequently been without apparent effect on the course of the infection. Studies of IV systems by CDC have shown a minimum of 6 percent prevalence of contamination within the tubing or bottles after the system has been in use. A significantly greater risk of contamination was noted in all systems where administration apparatus remained unchanged for greater than 48 hours. The studies also revealed that routine once-daily complete change of all IV administration apparatus, especially at the time of replacement of infusion devices (polyethylene catheters, needles, etc.) can greatly decrease the hazard of extrinsic contamination by preventing introduced organisms from propagating to dangerous levels. Bacterial contamination of the outer surface of the insert discs (synthetic cap liner) of unopened Abbott bottles has recently been demonstrated by CDC, which ranges from 0 to 52 percent among sampled lots. Bacillis species, S. epidermidis, Pseudomonas maltophillia and yeasts have been most frequently isolated, however, E. cloacae or Erwinia species have been isolated from 12 of 212 caps tested. Between April and September 1970, Abbott Laboratories partially converted to a new type of cap liner. Direct sampling of fluids from intact non-manipulated bottles by CDC has been negative, but transfer of organisms from contaminated caps to the fluid has been effected approximately 25 percent of the time by sequentially striking the cap several times, unscrewing and replacing it, and then hanging the bottle inverted for 24 to 48 hours. Transfer of organisms from contaminated caps to the fluid of bottles, where the cap has not been manipulated, has not been demonstrated, but is currently under further investigation. Once inoculated into commercial dextrose containing solutions, organisms of the Klebsiella-enterobacter group are capable of proliferating at room temperature, whereas other tested members of the Enterobacteriacae or Staphylococci either fail to grow or die. (Reported by the Food and Drug Administration and the Center for Disease Control.) Reference:
Editorial Comment:The following press release was issued March 13, 1971: The Commissioner of Food and Drugs, Dr. Charles C. Edwards, and the Director of the Center for Disease Control, Dr. David J. Sencer, today announced that special precautions must be taken in hospitals, nursing homes, and other health care facilities to reduce the risk of septicemia from the use of Abbott Laboratories intravenous (IV) infusion products. While contamination resulting in septicemia can occur in the use of infusion products from any manufacturer, recent Abbott production appears to present a unique problem. These products will be replaced as rapidly as possible by Abbott, however, these solutions are essential for patient care and cannot be withdrawn before replacement is in hand. A rising incidence of septicemia caused by organisms rarely associated with septicemia has been found in connection with the use of intravenous fluids in eight hospitals surveyed by the CDC. All eight were users of the Abbott infusion system. CDC has been closely examining the fluids, the infusion apparatus, and clinical reports of septicemia. The plastic liners in some Abbott bottle caps have been found contaminated by the implicated organisms. A tentative conclusion is that the organisms can enter the fluid from the plastic cap liners when the caps are opened and replaced while the bottle is held for later use. There is no evidence that the closure system allows or contributes to contamination before it is opened. It has been shown that when the cap has been removed and replaced that migration of bacterial organism from the cap lining may occur. The bottles of fluid known to be involved have been manufactured from February 1970 and they bear codes beginning with 842 through 855. Teams of experts from CDC, FDA, and Abbott Labs are reviewing all aspects of the problem. This review will be completed within a few days and it is expected that a resolution will be developed rapidly. Meanwhile, with cooperation of the American Hospital Association, hospitals and other users of these solutions are being advised of special procedures to reduce the contamination hazard to a minimum. These procedures include: opening the containers at the point of use only; not replacing the cap; and using the contents of the containers immediately upon opening. Hospitals also are being advised to change IV apparatus at least every 24 hours. The CDC studies have demonstrated that any brand of IV apparatus is more likely to cause infection if left in place longer. CDC and FDA conclude that the joint actions being taken are reasonable and necessary in the interest of patient care and to prevent a disruption in the availability of these essential drugs. On the basis of the studies conducted thus far, several additional specific measures which might minimize the risk of contamination from Abbott products are recommended:
Editorial Note -- 1997Frank S Rhame, MD, Abbott Northwestern Hospital, Minneapolis; Dennis G Maki, MD, Professor of Medicine, University of Wisconsin, Madison; former EIS officers in the Hospital Infections Section, Bacterial Diseases Branch, CDC, John V Bennett, MD, former Director, Division of Bacterial Diseases, CDC; and William R Jarvis, MD, Acting Director, Hospital Infections Program, National Center for Infectious Diseases, CDC. In December 1970, CDC's Hospital Infections Section, Bacterial Diseases Branch, first received reports of episodes of nosocomial Enterobacter sp. bloodstream infection (BSI). There was early concern about an association with receipt of intravenous (IV) fluids because the patients had no primary infections or cultures yielding Enterobacter. Initially, it was hypothesized that this was caused by the vulnerability of IV solution bottle screw-cap closures to extrinsic contamination. By January 1971, several hospitals across the United States reported patients with BSIs caused by E. cloacae and an organism then uncharacterized as a human pathogen, E. agglomerans. Furthermore, many of the episodes occurred in patients more healthy than those who ordinarily were at risk for nosocomial BSIs, and all of the hospitals used IV fluids manufactured by one company, Abbott Laboratories, Inc. On-site epidemiologic investigations were initiated at two hospitals by Epidemic Intelligence Service (EIS) officers from CDC's Hospital Infection Section. The investigations could not relate the IV-associated BSIs at these hospitals to a particular additive or to any IV system component and could not identify any defect in IV system management leading to extrinsic contamination. In general, contamination outbreaks involve a single organism; in these investigations, two bacterial species were involved. Nevertheless, the finding of these same two unusual organisms causing BSIs at multiple hospitals decreased the likelihood of extrinsic contamination and raised the possibility of a common source. By February 1971, epidemic organisms were found in the caps of IV solution bottles. Opened and unopened bottles of IV solution were obtained, and cultures of these IV fluid bottles were initiated at CDC. In vitro studies documented that simple, commonly performed manipulations, such as unscrewing the cap, adding a medication, replacing the cap, and inverting the bottle for mixing would transfer the contaminating organisms from the cap to the fluid. After the warning published in MMWR on March 6, 1971, CDC and Food and Drug Administration (FDA) teams conducted investigations at both of the Abbott manufacturing plants (1). Ultimately, CDC isolated the epidemic strains directly from the solutions in 13 (0.7%) of 1825 1-liter bottles (2). By March 1971, E. cloacae and E. agglomerans BSIs had been reported from eight hospitals across the United States. Approximately 20 persons at CDC were involved in assembling data from these hospitals, culturing IV bottles and working with Abbott and FDA on the appropriate response to the situation. The bases for action were the occurrence of unexpected primary nosocomial BSIs in multiple hospitals exclusively among patients receiving Abbott IV solutions, the recovery of epidemic organism from bottle caps and several hanging IV bottles, and the termination of an outbreak in a hospital after changing from Abbott IV fluids. Meetings in Washington, D.C., led to the decision to issue the joint press release on March 13 by Charles Edwards, M.D., Commissioner of Food and Drugs, and David Sencer, M.D., Director of CDC. The need for an MMWR special supplement, without precedent for this publication, arose because the MMWR dated March 13 had been finalized by Wednesday, March 11. The decision to issue the press release was not made until Thursday and the data under-lying it could not have been added to the MMWR dated March 13. The March 13 governmental action was not a total recall of all Abbott IV fluids because it was not clear that there was sufficient substitute IV solution from alternate manufacturers to meet the nation's needs. To reduce the risk for BSI, instructions on proper IV bottle manipulation were issued to minimize transfer of organisms from the cap to the fluid. On March 22, FDA issued a recall of all Abbott IV fluids when it became clear that the scope of the epidemic was larger than initially recognized, that some hospitals continued to have episodes of BSI despite implementation of the March 13 recommendations, and that sufficient substitute IV fluids were available (3). By March 31, a total of 405 patients with BSI had been reported, of whom none had had onset after the March 22 recall (1). The full epidemiologic analysis published in 1976 (4 ) was delayed in part because of medicolegal considerations, and underscored the exceptional magnitude of the outbreak: estimates of the magnitude ranged from 2000 to 8000 episodes of BSI caused by these contaminated IV fluids. Approximately 10% of the case-patients in the studied hospitals died while bacteremic or soon thereafter. The cause of this outbreak was ultimately attributed to unexpected consequences of a new cap design for IV solution bottles (1). After the March 13 warning, it was learned that the company had been phasing in a new cap type since approximately March 1970, although substantial production did not occur until about November 1970. The old cap liner incorporated a natural red rubber disc between the metal cap and a thin Gilsonite‰ wafer that was pressed against the bottle orifice by the natural rubber. Fortuitously, the rubber had antibacterial properties against E. agglomerans and E. cloacae, two organisms unusual among bacteria in their ability to proliferate in acidic, nitrogen-poor, dextrose-containing fluids (5). In vitro studies showed that these organisms were drawn up into the cap as cooling occurred after autoclaving but were not destroyed by the new elastomer lined cap. Manufacturing plant environmental cultures documented that these organisms were abundant in the manufacturing plant environment because of spillage of glucose-containing solution that favored their growth. It is uncertain whether the organisms may have been drawn into the fluid itself during cooling or whether it was impossible to remove the caps without transferring organism to the fluid. Furthermore, the United States Pharmacopeia (USP) sampling procedures did not require identification of contaminating organisms, and the required two-step sampling scheme was too insensitive -- missing 98% of lots contaminated at a 1% rate. Identification of recovered organisms would probably have revealed the problem earlier. Surveillance of nosocomial infections had begun at CDC in 1965 when the Hospital Infection Section initiated the Comprehensive Hospital Infections Program (CHIP) study in six community hospitals. CHIP produced high-quality data on infection rates in hospitalized patients and was used to develop nosocomial infection surveillance methods applicable to U.S. community hospitals. In May 1969, CDC initiated the National Nosocomial Infections Surveillance (NNIS) system, a substantially larger program that accepted data from up to 80 hospitals nationwide. These efforts helped establish nosocomial infection surveillance programs in U.S. hospitals. Without such programs, the detection of this outbreak would have been more difficult, and the outbreak likely would have resulted in additional deaths. While several hospitals recognized the phenomenon independently because of clustering of BSIs and/or by the involvement of the unusual organisms, most involved hospitals had small numbers of infected patients and recognized their involvement only in retrospect (6). This outbreak required a compilation of national prospective data to detect, investigate, and terminate. The epidemiologic expertise and laboratory resources required to carry out multiple onsite investigations and in vitro microbiologic studies in a timely fashion were only available at CDC, a national public health institution. During February-April 1971, approximately 3000 cultures of large volume parenteral fluids were performed by CDC laboratory personnel -- the extraordinarily large number of cultures resulted in a national shortage of brain heart infusion broth. The necessary and synergistic interaction between epidemiologists and microbiologists in solving the outbreak is still recognized by the prestigious Mackel Award, offered annually to EIS officers in honor of the competence and dedication of the late Donald C. Mackel, who directed the laboratory studies. This outbreak also resulted in development of more sensitive and restrictive USP requirements for monitoring contamination of large volume parenterals by manufacturers and the development of the first guidelines by CDC on the prevention of nosocomial infection (7,8). These guidelines were the precursors to the current Hospital Infections Program Nosocomial Infection Prevention Guidelines. One consequence of this outbreak was the media attention and litigation incurred by some of the involved hospitals. Ironically, it was the active surveillance for nosocomial infections at these hospitals and the quality of their infection-control programs that facilitated recognition of this outbreak. Headlines about BSIs in hospitalized patients were not mitigated by the hospitals' ultimate medicolegal exoneration. The manufacturer incurred considerable economic consequence from this outbreak, even though this outbreak occurred before the era of cooperation between plaintiff's attorneys in product liability suits. The trials were conducted in 1976, and most of the epidemiologic evidence was not admitted. Judgments turned on whether an individual lot could be demonstrated to be contaminated and whether the patient could be proven to have received fluid from that lot. Since lot numbers were not often recorded, few plaintiffs prevailed. One of the special features of the CDC in response to this crisis was the compression of hierarchy, including the open information channels throughout CDC. For example, during the most intense period of the investigation, the CDC Director was in frequent direct conversation with front-line EIS officers. In negotiating sessions with FDA in Washington, data presentations were made by EIS officers. This large nationwide outbreak of nosocomial BSIs traced to intrinsic contamination of IV solutions led to widespread changes at industry, hospital, state, and federal levels. Expansion of nosocomial infection surveillance and control programs occurred at the hospital and federal level. There was enhancement of FDA and CDC surveillance for outbreaks attributable to potentially contaminated products, expansion of training programs for infection-control professionals and hospital epidemiologists, development of guidelines for the prevention of nosocomial infection, strengthening of CDC core epidemiologic and laboratory capacity to respond to nationwide outbreaks, and strengthening of FDA and USP requirements for monitoring potential product contamination. Since the institution of these measures, no large nationwide outbreak of BSIs traced to intrinsically contaminated IV solutions has occurred in the United States. References
Original report published with new editorial note in MMWR 1997;46:1227-33 (December 26, 1997). Figure_1 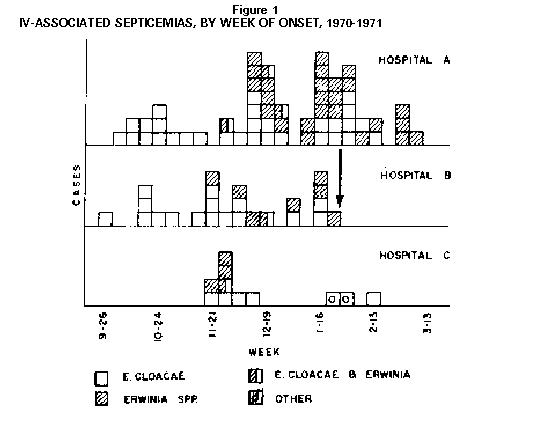 Return to top. Disclaimer All MMWR HTML versions of articles are electronic conversions from ASCII text into HTML. This conversion may have resulted in character translation or format errors in the HTML version. Users should not rely on this HTML document, but are referred to the electronic PDF version and/or the original MMWR paper copy for the official text, figures, and tables. An original paper copy of this issue can be obtained from the Superintendent of Documents, U.S. Government Printing Office (GPO), Washington, DC 20402-9371; telephone: (202) 512-1800. Contact GPO for current prices. **Questions or messages regarding errors in formatting should be addressed to mmwrq@cdc.gov.Page converted: 04/20/99 |
|||||||||
This page last reviewed 5/2/01
|
{kind=link}