 |
|
|
|
|
|
|
| ||||||||||
|
|
|
|
|
|
|
||||
| ||||||||||
|
|
|
|
|
Persons using assistive technology might not be able to fully access information in this file. For assistance, please send e-mail to: mmwrq@cdc.gov. Type 508 Accommodation and the title of the report in the subject line of e-mail. Prevention of Rotavirus Gastroenteritis Among Infants and Children Recommendations of the Advisory Committee on Immunization Practices (ACIP)Please note: An erratum has been published for this article. To view the erratum, please click here.Prepared by
The material in this report originated in the National Center for Immunization and Respiratory Diseases, Anne Schuchat, MD, Director, and the Division of Viral Diseases, Larry Anderson, MD, Director. Corresponding preparer: Margaret M. Cortese, MD, National Center for Immunization and Respiratory Diseases, CDC, 1600 Clifton Rd., NE, MS A-47, Atlanta GA 30333. Telephone: 404-639-1929; Fax: 404-639-8665; E-mail: mcortese@cdc.gov. SummaryRotavirus is the most common cause of severe gastroenteritis in infants and young children worldwide. Before initiation of the rotavirus vaccination program in the United States in 2006, approximately 80% of U.S. children had rotavirus gastroenteritis by age 5 years. Each year during the 1990s and early 2000s, rotavirus resulted in approximately 410,000 physician visits, 205,000-272,000 emergency department visits, and 55,000-70,000 hospitalizations among U.S. infants and children, with total annual direct and indirect costs of approximately $1 billion. In February 2006, a live, oral, human-bovine reassortant rotavirus vaccine (RotaTeq® [RV5]) was licensed as a 3-dose series for use among U.S. infants for the prevention of rotavirus gastroenteritis, and the Advisory Committee on Immunization Practices (ACIP) recommended routine use of RV5 among U.S. infants (CDC. Prevention of rotavirus gastroenteritis among infants and children: recommendations of the Advisory Committee on Immunization Practices [ACIP]. MMWR 2006;55[No. RR-12]). In April 2008, a live, oral, human attenuated rotavirus vaccine (Rotarix® [RV1]) was licensed as a 2-dose series for use among U.S. infants, and in June 2008, ACIP updated its rotavirus vaccine recommendations to include use of RV1. This report updates and replaces the 2006 ACIP statement for prevention of rotavirus gastroenteritis. ACIP recommends routine vaccination of U.S. infants with rotavirus vaccine. RV5 and RV1 differ in composition and schedule of administration. RV5 is to be administered orally in a 3-dose series, with doses administered at ages 2, 4, and 6 months. RV1 is to be administered orally in a 2-dose series, with doses administered at ages 2 and 4 months. ACIP does not express a preference for either RV5 or RV1. The recommendations in this report also address the maximum ages for doses, contraindications, precautions, and special situations for the administration of rotavirus vaccine. IntroductionRotavirus is the most common cause of severe gastroenteritis in infants and young children worldwide. Rotavirus causes approximately half a million deaths each year among children aged <5 years, with >80% of deaths occurring in developing countries (1). In the United States during the prevaccine era, rotavirus gastroenteritis resulted in relatively few childhood deaths (approximately 20-60 deaths per year among children aged <5 years) (2--5). However, before initiation of the rotavirus vaccination program in 2006, nearly every child in the United States was infected with rotavirus by age 5 years; the majority had gastroenteritis, resulting annually during the 1990s and early 2000s in approximately 410,000 physician visits, 205,000-272,000 emergency department (ED) visits, 55,000-70,000 hospitalizations, and total annual direct and indirect costs of approximately $1 billion (5--9) (Figure 1). This report presents the recommendations of the Advisory Committee on Immunization Practices (ACIP) for use of two rotavirus vaccines among U.S. infants: RotaTeq® (RV5) (Merck and Company, Whitehouse Station, New Jersey), which was licensed by the Food and Drug Administration (FDA) in February 2006 (10) and Rotarix® (RV1) (GlaxoSmithKline [GSK] Biologicals, Rixensart, Belgium), which was licensed by FDA in April 2008 (11). This report updates and replaces the 2006 ACIP statement for prevention of rotavirus gastroenteritis (12). BackgroundClinical and Epidemiologic Features of Rotavirus Disease in the Prevaccine Era In the prevaccine era, rotavirus infected almost all children by age 5 years; severe dehydrating gastroenteritis caused by rotavirus occurred primarily among children aged 4-23 months (13--15). Rotavirus infects the proximal small intestine, where it elaborates an enterotoxin and destroys the epithelial surface, resulting in blunted villi, extensive damage, and shedding of massive quantities of virus in stool (13). The estimated incubation period for rotavirus diarrheal illness is <48 hours (16). Under experimental conditions, adults who became ill had symptoms 1--4 days after receiving rotavirus orally (17,18). The clinical spectrum of rotavirus illness in children ranges from mild, watery diarrhea of limited duration to severe diarrhea with vomiting and fever than can result in dehydration with shock, electrolyte imbalance, and death (19). The illness usually begins with acute onset of fever and vomiting, followed 24--48 hours later by frequent, watery stools (20,21). Up to one third of children with rotavirus illness have a temperature of >102ºF (>39ºC) (22,23). Vomiting usually lasts <24 hours; other gastrointestinal symptoms generally resolve in 3-7 days. Rotavirus protein and ribonucleic acid (RNA) have been detected in blood, organs, and cerebrospinal fluid, but the clinical implications of these findings are not clear (20,24). Rotaviruses are shed in high concentrations (i.e., 1012 virus particles per gram of stool during the acute illness) in the stools of infected children before and several days after clinical disease (25). Rotavirus is transmitted primarily by the fecal-oral route, both through close person-to-person contact and through fomites (26). Very few infectious virions are needed to cause disease in susceptible hosts (25). Spread is common within families. Of adult contacts of infected children, 30%-50% become infected, although infections in adults often are asymptomatic because of immunity from previous exposure (27--29). Transmission of rotavirus through contaminated water or food is likely to be rare (30,31). Transmission through airborne droplets also has been hypothesized but remains unproven (21,30,32). In the United States, rotavirus causes winter seasonal peaks of gastroenteritis, with activity beginning usually in the southwestern states during December-January, moving across the country, and ending in the northeastern states in April-May (33--35). Rotavirus might account for up to 10% of gastroenteritis episodes among children aged <5 years (36). Infants and children with rotavirus gastroenteritis are likely to have more severe symptoms than those with nonrotavirus gastroenteritis (22,23,37,38). In the prevaccine era, rotavirus accounted for 30%-50% of all hospitalizations for gastroenteritis among U.S. children aged <5 years and up to 70% of hospitalizations for gastroenteritis during the seasonal peak months (7,14,39--44). Of all the rotavirus hospitalizations that occurred among children aged <5 years in the United States during the prevaccine era, 17% occurred during the first 6 months of life, 40% by age 1 year, and 75% by age 2 years (Figure 2). Rotavirus accounted for 20%--40% of outpatient clinic visits during the rotavirus season (14,45,46). Before the initiation of the rotavirus vaccination program, four of five children in the United States had rotavirus gastroenteritis by age 5 years (36,39,47), one in seven required a clinic or ED visit, one in 70 were hospitalized, and one in 200,000 died from this disease (3,8). Active, population-based surveillance from early 2006 and before vaccine was used provided annual rotavirus hospitalization and ED visit rates of 22.4 and 301 per 10,000 children aged <3 years, respectively (14). Rotavirus also was an important cause of hospital-acquired gastroenteritis among children (48). In a recent study, factors associated with increased risk for hospitalization for rotavirus gastroenteritis among U.S. children included lack of breastfeeding, low birth weight (a likely proxy for prematurity), daycare attendance, the presence of another child aged <24 months in the household, and either having Medicaid insurance or having no medical insurance (49). Another study identified low birth weight, maternal factors (e.g., young age, having Medicaid insurance, and maternal smoking), and male gender as risk factors for hospitalization with viral gastroenteritis (50). These studies suggest that preterm infants are at higher risk for severe rotavirus disease. Children and adults who are immunocompromised because of congenital immunodeficiency or because of bone marrow or solid organ transplantation sometimes experience severe or prolonged rotavirus gastroenteritis (51--56). The severity of rotavirus disease among children infected with human immunodeficiency virus (HIV) might be similar to that among children without HIV infection (57). Whether the incidence rate of severe rotavirus disease among HIV-infected children is similar to or greater than that among children without HIV infection is not known. Laboratory Testing for RotavirusBecause the clinical features of rotavirus gastroenteritis do not differ distinctly from those of gastroenteritis caused by other pathogens, confirmation of rotavirus infection by laboratory testing of fecal specimens is necessary for reliable rotavirus surveillance and can be useful (e.g., for infection-control purposes) in clinical settings. The most widely used diagnostic laboratory method is antigen detection in the stool by an enzyme immunoassay (EIA) directed at an antigen common to all group A rotaviruses (i.e., those that are the principal cause of human disease). Certain commercial EIA kits are available that are easy to use, rapid, and highly sensitive, making them suitable for rotavirus surveillance and clinical diagnosis. Other techniques, including electron microscopy, RNA electrophoresis, reverse transcription--polymerase chain reaction (RT-PCR), sequence analysis, and culture are used primarily in research settings. Serologic methods that detect a rise in serum antibodies, primarily EIA for rotavirus serum immunoglobulin G (IgG) and immunoglobulin A (IgA) antibodies, have been used to confirm recent infections primarily in the research setting. In vaccine trials, the immunogenicity of rotavirus vaccines has been assessed by measuring rotavirus-specific IgG, IgA and neutralizing antibodies to the serotypes of the vaccine strains (58--60). Morphology, Antigen Composition, and Immune ResponseRotaviruses are 70-nm nonenveloped RNA viruses in the family Reoviridae (61,62). The viral nucleocapsid is composed of three concentric shells that enclose 11 segments of double-stranded RNA. The outermost layer contains two structural viral proteins (VP): VP4, the protease-cleaved protein (P protein) and VP7, the glycoprotein (G protein). These two proteins define the serotype of the virus and are considered critical to vaccine development because they are targets for neutralizing antibodies that are believed to be important for protection (61,62). Because the two gene segments that encode these proteins can segregate independently, a typing system consisting of both P and G types has been developed (63). Although characterizing G serotypes by traditional methods is straightforward, using these methods for determining P serotypes is more difficult. Consequently, molecular methods are used almost exclusively to define genetically distinct P genotypes by nucleotide sequencing. These genotypes correlate well with known serotypes, but they are designated in brackets (e.g., P[8]) to distinguish them from P serotypes determined by antigenic analyses. In the United States, viruses containing six distinct P and G combinations are most prevalent: P[8]G1, P[4]G2, P[8]G3, P[8]G4, P[8]G9, P[6]G9 (64--67) (Figure 3). Several animal species (e.g., primates and cows) are susceptible to rotavirus infection and suffer from rotavirus diarrhea, but animal strains of rotavirus differ from those that infect humans. Although human rotavirus strains that possess a high degree of genetic homology with animal strains have been identified (63,68--71), animal-to-human transmission appears to be uncommon. However, natural reassortant animal-human strains have been identified in humans (63), and some are being developed as vaccine candidates (72). Although children can be infected with rotavirus several times during their lives, initial infection after age 3 months is most likely to cause severe gastroenteritis and dehydration (15,73--75). After a single natural infection, 38% of children are protected against subsequent infection with rotavirus, 77% are protected against subsequent rotavirus gastroenteritis, and 87% are protected against severe rotavirus gastroenteritis; second and third infections confer progressively greater protection against rotavirus gastroenteritis (75). Rotavirus infection in healthy full-term neonates often is asymptomatic or results in only mild disease, perhaps because of protection from passively transferred maternal antibody (13,76). The immune correlates of protection from rotavirus infection and disease are not understood fully. Both serum and mucosal antibodies probably are associated with protection, and in some studies, serum antibodies against VP7 and VP4 have correlated with protection (58,59). However, in other studies, including vaccine studies, correlation between serum antibody and protection has been poor (77). First infections with rotavirus generally elicit a predominantly homotypic, serum-neutralizing antibody response, and subsequent infections typically elicit a broader, heterotypic response (21,78). The influence of cell-mediated immunity is understood less clearly but probably is related both to recovery from infection and to protection against subsequent disease (79,80). Rotavirus VaccinesBackground In 1998, ACIP recommended Rotashield® (RRV-TV) (Wyeth Lederle Vaccines and Pediatrics, Marietta, Pennsylvania) (81), a rhesus-based tetravalent rotavirus vaccine, for routine vaccination of U.S. infants, with 3 doses administered at ages 2, 4, and 6 months (82). However, RRV-TV was withdrawn from the U.S. market within 1 year of its introduction because of its association with intussusception (83). At the time of its withdrawal, RRV-TV had not yet been introduced in any other national vaccination program globally. The risk for intussusception was most elevated (>20-fold increase) within 3-14 days after receipt of dose 1 of RRV-TV, with a smaller (approximately fivefold) increase in risk within 3-14 days after receipt of dose 2 (84). Overall, the estimated risk associated with dose 1 of RRV-TV was approximately one case per 10,000 vaccine recipients (85). After they reassessed the data on RRV-TV and intussusception, certain researchers suggested that the risk for intussusception was age-dependent and that the absolute number of intussusception events, and possibly the relative risk for intussusception associated with dose 1 of RRV-TV increased with increasing age at vaccination (86,87). However, after reviewing all the available data, the World Health Organization (WHO) Global Advisory Committee on Vaccine Safety (GACVS) concluded that the risk for RRV-TV--associated intussusception was high in infants vaccinated after age 60 days and that insufficient evidence was available to conclude that the use of RRV-TV at age <60 days was associated with a lower risk (88). GACVS noted that the possibility of an age-dependent risk for intussusception should be taken into account in assessing rotavirus vaccines. MethodologyThe ACIP rotavirus vaccine workgroup was reestablished in July 2007, after submission of the Biologics License Application (BLA) for RV1 to FDA in June 2007. The workgroup held teleconferences at least monthly to review published and unpublished data on the burden and epidemiology of rotavirus disease in the United States, the safety and efficacy of RV1 and RV5, and cost-effectiveness analyses. Recommendation options were developed and discussed by ACIP's rotavirus vaccine work group. The opinions of workgroup members and other experts were considered when data were lacking. Programmatic aspects related to implementation of the recommendations were taken into account. Presentations were made to ACIP during meetings in October 2007 and February 2008. The final proposed recommendations were presented to ACIP at the June 2008 ACIP meeting; after discussion, minor modifications were made, and the recommendations were approved. Pentavalent Human-Bovine Reassortant Rotavirus Vaccine (RotaTeq® [RV5])RV5, which was licensed in the United States in 2006, is a live, oral vaccine that contains five reassortant rotaviruses developed from human and bovine parent rotavirus strains (Box) (10,89). Four reassortant rotaviruses express one of the outer capsid proteins (G1, G2, G3, or G4) from the human rotavirus parent strains and the attachment protein (P7[5]) from the bovine rotavirus parent strain. The fifth reassortant virus expresses the attachment protein (P1A[8]) from the human rotavirus parent strain and the outer capsid protein (G6) from the bovine rotavirus parent strain. The parent bovine rotavirus strain, Wistar Calf 3 (WC3), was isolated in 1981 from a calf with diarrhea in Chester County, Pennsylvania, and was passaged 12 times in African green monkey kidney cells (90). The reassortants are propagated in Vero cells using standard tissue culture techniques in the absence of antifungal agents. The licensed vaccine is a ready-to-use 2 ml solution that contains >2.0-2.8 x 106 infectious units (IUs) per individual reassortant dose, depending on serotype. The RV5 BLA contained three phase III trials (91). Data from these trials on the immunogenicity, efficacy, and safety of RV5 are summarized below. ImmunogenicityA relation between antibody responses to rotavirus vaccination and protection against rotavirus gastroenteritis has not been established. In clinical trials, a rise in titer of rotavirus group-specific serum IgA antibodies was used as one of the measures of the immunogenicity of RV5. Sera were collected before vaccination and at 2--6 weeks after dose 3, and seroconversion was defined as a threefold or greater rise in antibody titer from baseline. Seroconversion rates for IgA antibody to rotavirus were 93%-100% among 439 RV5 recipients compared with 12%-20% in 397 placebo recipients in phase III studies (91). Antibody responses to concomitantly administered vaccines were evaluated in a study with a total of 662 RV5 recipients and 696 placebo recipients. Different subsets of infants were evaluated for specific antibody responses. A 3-dose series of RV5 did not diminish the immune response to concomitantly administered Haemophilus influenzae type b conjugate (Hib) vaccine, inactivated poliovirus vaccine (IPV), hepatitis B (HepB) vaccine, pneumococcal conjugate vaccine (PCV), and diphtheria and tetanus toxoids and acellular pertussis (DTaP) vaccine (10,91). EfficacyThe efficacy of the final formulation of RV5 has been evaluated in two phase III trials among healthy infants (92,93). Administration of oral polio vaccine (OPV) was not allowed; concomitant administration of other vaccines was not restricted. The large Rotavirus Efficacy and Safety Trial (REST) included a clinical efficacy substudy (Tables 1 and 2). In this substudy, 4,512 infants from Finland and the United States were included in the primary per-protocol efficacy analysis (consisting of evaluable subjects for whom there was no protocol violation) through one rotavirus season. The primary efficacy endpoint was the prevention of wild type G1-G4 rotavirus gastroenteritis occurring >14 days after completion of a 3-dose series through the first full rotavirus season after vaccination. A case of rotavirus gastroenteritis was defined as production of three or more watery or looser-than-normal stools within a 24-hour period or forceful vomiting, along with rotavirus detection by EIA in a stool specimen obtained within 14 days after the onset of symptoms. G serotypes were identified by RT-PCR followed by sequencing. Severe gastroenteritis was defined as a score of >16 on an established 24-point severity scoring system (Clark score) on the basis of intensity and duration of fever, vomiting, diarrhea, and changes in behavior. The efficacy of RV5 against G1-G4 rotavirus gastroenteritis of any grade of severity through the first full rotavirus season after vaccination was 74.0% (95% confidence interval [CI] = 66.8-79.9) and against severe G1-G4 rotavirus gastroenteritis was 98.0% (CI = 88.3-100.0) (Table 2). RV5 reduced office or clinic visits for G1-G4 rotavirus gastroenteritis by 86.0% (CI = 73.9-92.5). In a trial that evaluated RV5 at the end of its shelf life, the efficacy estimates for RV5 based on per-protocol analysis of data from 551 RV5 recipients and 564 placebo recipients were similar to those identified in the clinical efficacy substudy (10,92,93). Among the limited number of infants from phase III trials who received at least 1 dose of RV5 (n = 144) or placebo (n = 135) >10 weeks after a previous dose, the estimate of efficacy of the RV5 series for protection against G1-G4 rotavirus gastroenteritis of any severity was 63% (CI = 53%--94%) (94). In the health-care utilization cohort of REST, data from 57,134 infants from 11 countries were included in the per-protocol analysis of the efficacy of RV5 in reducing the need for hospitalization or ED care for rotavirus gastroenteritis (93). The efficacy of the RV5 series against ED visits for G1-G4 rotavirus gastroenteritis was 93.7% (CI = 88.8-96.5), and efficacy against hospitalization for G1-G4 rotavirus gastroenteritis was 95.8% (CI = 90.5-98.2) (Table 2). Efficacy was observed against all G1-G4 and G9 serotypes (Table 3); relatively few non-G1 rotavirus cases were detected. The efficacy of RV5 against all gastroenteritis-related hospitalizations was 58.9% (CI = 51.7-65.0) for the period that started after dose 1. Breastfeeding did not appear to diminish the efficacy of a 3-dose series of RV5. Post-hoc analyses of the clinical efficacy substudy found that the efficacy of RV5 against G1-G4 rotavirus gastroenteritis of any severity through the first rotavirus season was similar among the 1,632 infants (815 in the vaccine group and 817 in the placebo group) who never were breastfed (68.3%; CI = 46.1-82.1) and the 1,566 infants (767 in the vaccine group and 799 in the placebo group) who were exclusively breastfed (68.0%; CI = 53.8--78.3) (95). Efficacy against severe G1-G4 rotavirus gastroenteritis also was similar among infants who never were breastfed (100.0%; CI = 48.3-100.0) and those who were exclusively breastfed (100.0%; CI = 79.3-100.0). In posthoc analyses of data from the clinical efficacy substudy of REST, efficacy also was estimated among 73 healthy preterm infants (gestational age of <37 weeks) who received RV5 and 78 healthy preterm infants who received placebo (96). The efficacy through the first full season against rotavirus gastroenteritis of any severity (all serotypes combined) was 73.0% (CI = -2.2--95.2); three cases occurred among RV5 recipients, and 11 cases occurred among placebo recipients. In the health- care utilization cohort, the efficacy against rotavirus gastroenteritis--attributable hospitalizations (all serotypes combined) for healthy preterm infants was 100.0% (CI = 53.0-100.0); no cases were identified among 764 preterm infants who received RV5 and nine cases were identified among 818 preterm infants who received placebo. Efficacy against rotavirus gastroenteritis--attributable ED visits was 100% (CI = 66.6-100.0), with no cases identified among RV5 recipients and 12 cases identified among placebo recipients (96). Adverse Events After VaccinationIntussusception REST was designed as a large trial to permit evaluation of safety with respect to intussusception; 69,625 enrolled infants received at least 1 dose of RV5 or placebo (10,93). No increased risk for intussusception was observed in this trial after administration of RV5 when compared with placebo. For the prespecified period of days 0-42 after any dose, six confirmed intussusception cases occurred among 34,837 infants who received RV5, and five confirmed intussusception cases occurred among 34,788 infants who received placebo (relative risk adjusted for group sequential design: 1.6; CI = 0.4-6.4). None of the infants with confirmed intussusception in either treatment group had onset during days 1--21 after dose 1. Other Adverse EventsSerious adverse events (SAEs) and deaths were evaluated in infants enrolled in phase III trials (10,97). Among RV5 and placebo recipients, the incidence of SAEs within 42 days of any dose (2.4% of 36,150 and 2.6% of 35,536, respectively) was similar. Across the studies, the incidence of death was similar among RV5 recipients (<0.1% [n = 25]) and placebo recipients (<0.1% [n = 27]). The most common cause of death (accounting for 17 ([32.7%]) of 52 deaths) was sudden infant death syndrome (SIDS), which was observed in eight RV5 recipients and nine placebo recipients. Gastroenteritis occurring anytime after a dose was reported as an SAE in 76 (0.2%) RV5 recipients and in 129 (0.4%) placebo recipients. Seizures reported as SAEs occurred in 27 (<0.1%) vaccine recipients and in 18 (<0.1%) placebo recipients (difference not statistically significant). Pneumonia occurring anytime after a dose was reported as an SAE in 59 (0.2%) of RV5 recipients and in 62 (0.2%) of placebo recipients; hospitalization for pneumonia within 7 days after any dose occurred in 11 (<0.1%) RV5 recipients and in 14 (<0.1%) placebo recipients (91). A subset of 11,711 infants was studied in detail to assess other potential adverse experiences (10). In the 42-day period postvaccination of any dose of RV5, the incidence of fever reported by parents and guardians of RV5 recipients and placebo recipients (42.6% and 42.8%, respectively) was similar, as was the incidence of hematochezia reported as an adverse experience (0.6% in both RV5 recipients and placebo recipients). Some (e.g., diarrhea, vomiting) adverse events occurred at a statistically higher incidence within 42 days of any dose in RV5 recipients (Table 4). Statistical significance was determined using 95% CIs on the risk difference; intervals with a lower bound above zero were considered statistically significant. Adverse events also were solicited from parents and guardians within the first week after each dose. RV5 recipients had a small but statistically significantly greater (p-value <0.05) rate of diarrhea and vomiting after specific doses or after any dose (Table 5). Among the limited number of infants from phase III trials who received at least 1 dose of RV5 or placebo >10 weeks after a previous dose (depending on dose number and specific adverse event monitored, the number of infants evaluated in either the RV5 or placebo group ranged from 211--1,182), the proportion of infants with adverse events appeared generally similar among the RV5 and placebo recipients (94). In the phase III clinical trials, infants were followed for up to 42 days of vaccine dose. Kawasaki disease was reported in five of 36,160 RV5 recipients and in one of 35,536 placebo recipients (unadjusted relative risk: 4.9; CI = 0.6--239.1) (10). Preterm InfantsIn posthoc analyses of data from REST, adverse events were examined among healthy preterm infants with gestational age of 25-36 weeks (median: 34 weeks) (10,96). At least one SAE was reported within 42 days after any dose in 55 (5.5%) of the 1,005 preterm infants who received RV5 and in 62 (5.8%) of the 1,061 preterm infants who received placebo. Among the preterm infants with gestational age of <32 weeks, at least one SAE was reported within 42 days of any dose in 6 (8.1%) of the 74 RV5 recipients and in 9 (9.8%) of the 92 placebo recipients. No confirmed intussusception occurred in a preterm infant during the study. Two deaths occurred in the RV5 group (one from SIDS and one from a motor-vehicle crash), and two occurred in the placebo group (one from SIDS and one from an unknown cause). The incidence of solicited adverse events (fever, vomiting, diarrhea, and irritability) within 7 days after each dose administration was assessed in preterm infants; depending on dose number and specific adverse event monitored, the number of infants evaluable in either the RV5 or placebo group varied (range: 108--154). The rates appeared generally similar between the RV5 and placebo recipients. Shedding and Transmission of Vaccine VirusFecal shedding of rotavirus vaccine virus was evaluated by plaque assays with electrophenotyping in a subset of infants enrolled in the large phase III trial by obtaining a single stool sample during days 4-6 after each dose of RV5 (93). Vaccine virus was detected in 17 (12.7%) of 134 infants after dose 1, zero of 109 infants after dose 2, and zero of 99 infants after dose 3. Shedding of vaccine virus also was assessed for phase III studies overall, including that detected by plaque assays of rotavirus-antigen positive stools from infants evaluated for possible gastroenteritis. Shedding was observed as early as 1 day and as late as 15 days after a dose (10). The potential for transmission of vaccine virus to other persons was not assessed. Postlicensure Rotavirus Surveillance Data from the United StatesRotavirus surveillance data from two systems, the National Respiratory and Enteric Virus Surveillance System (NREVSS) and the New Vaccine Surveillance Network (NVSN), indicated that the 2007--08 season was substantially delayed in onset and diminished in magnitude compared to the seasons before substantial uptake of RV5 among U.S. infants (98). NREVSS is a voluntary network of U.S laboratories that provides CDC with weekly reports of the number of tests performed and positive results obtained for a variety of pathogens. For rotavirus, results of EIAs are reported. Compared with the 15 previous seasons spanning 1991-2006, rotavirus activity during the 2007-08 season appeared delayed in onset by 2-4 months (Figure 4). Further, data from the 32 laboratories that consistently reported results during July 2000--May 2008 indicated that the number of tests positive for rotavirus during the 2007--08 season (January 1, 2008--May 3, 2008) was lower by more than two thirds compared with the median number positive during the same weeks in the seven preceding rotavirus seasons. Since 2006, NVSN has conducted prospective, population-based surveillance for rotavirus gastroenteritis among children aged <3 years residing in three U.S counties. Among children with gastroenteritis enrolled during January--April of each year, the overall percentage of fecal specimens testing positive for rotavirus was 51% in 2006, 54% in 2007, and 6% in 2008. Although nationally representative data on vaccine coverage are not yet available, information from population-based immunization information system sentinel sites indicates that mean coverage with 1 dose of rotavirus vaccine among infants aged 3 months was 49.1% in May 2007 and 56.0% in March 2008. Additional surveillance and epidemiologic studies are underway to monitor the impact of rotavirus vaccination in the United States. Postlicensure Safety Monitoring Data from the United StatesDuring February 2006-March 2008, approximately 14 million doses of RV5 were distributed in the United States (99). Results from two safety monitoring systems have been reported. The U.S. Vaccine Adverse Event Reporting System (VAERS), a national passive surveillance system managed jointly by FDA and CDC, receives reports of adverse events after vaccination from multiple sources, including health-care providers, vaccine recipients and parents and guardians of vaccine recipients, and manufacturers (100,101). Reported cases of intussusception among vaccine recipients are classified as confirmed if Brighton Collaboration Level 1 criteria are met (102). In VAERS analyses, the number of confirmed intussusception cases reported after vaccination is compared with the number of cases expected to occur by chance alone. This latter number is determined from estimates of the background rates of intussusception among infants and estimates of the total number of doses of RV5 that have been administered to infants. As of March 31, 2008, the number of confirmed cases of intussusception reported to VAERS during either the 1--21 day period or the 1--7 day period after receipt of any dose (doses 1, 2, and 3 combined) of RV5 did not exceed the number of cases expected to occur by chance alone after vaccination (99,103). A relative increase in intussusception reports in the first week after receipt of dose 1 of RV5, compared with the second and third weeks after dose 1, has been noted; whether this phenomenon is related to better reporting for intussusception during the first week after vaccination or represents a small increased risk for intussusception during the first week after dose 1 of RV5 is not clear (99,103). Because VAERS is not designed to provide a definitive assessment of risk, the safety of RV5 also is monitored in the Vaccine Safety Datalink (VSD), a collaborative project between CDC and several large U.S. health maintenance organizations that links computerized patient-level vaccination data to medical outcomes, including potential adverse events (104). VSD is able to test hypotheses suggested by VAERS reports and prelicensure trials. With >200,000 doses of RV5 administered to infants in the VSD system during May 21, 2006--May 24, 2008, the number of cases of intussusception identified that occurred within a 30-day period after receipt of any dose of RV5 was not greater than the number of cases expected to occur by chance alone (105). No case of intussusception was identified that occurred within the first week after receipt of the first dose of RV5 in VSD (out of approximately 77,000 first doses) nor in the prelicensure REST. The data suggest that, if any associated risk exists, the risk for intussusception associated with the first dose of RV5 within the first week after vaccination is not greater than one in 25,000--50,000 first doses (105). Other adverse events monitored in VAERS, VSD, or both include hematochezia, Kawasaki syndrome, seizures, meningitis and encephalitis, myocarditis and gram-negative sepsis. The data do not indicate that RV5 is associated with an increased risk for these adverse events (99,105). Monovalent Human Rotavirus Vaccine (Rotarix® [RV1])RV1 is a live, oral vaccine licensed in 2008 for use in the United States that contains a human rotavirus strain (type G1P1A[8]) (Box. It was developed from a strain of rotavirus (termed 89-12) that was isolated in 1988 from a child in Cincinnati, Ohio, and that was first attenuated by passaging 33 times in African green monkey kidney cells (106); it was then cloned and further passaged in a Vero cell line and renamed RIX 4414 (107). The licensed vaccine is prepared as a lyophilized powder that is reconstituted with 1 ml of a calcium bicarbonate buffer to a titer of >106.0 CCID50 per dose (11). The BLA contained six phase II trials and five phase III trials (108). Data from these trials on the immunogenicity, efficacy, and safety of RV1 are summarized below. ImmunogenicityA relation between antibody responses to rotavirus vaccination and protection against rotavirus gastroenteritis has not been established. In two clinical trials, seroconversion was defined as the appearance of antirotavirus IgA antibodies (concentration of >20 U/ml) postvaccination in the serum of infants previously negative for rotavirus IgA antibodies. In the two studies, 1-2 months after a 2-dose series, 681 (86.5%) of 787 RV1 recipients seroconverted compared with 28 (6.7%) of 420 placebo recipients, and 302 (76.8%) of 393 RV1 recipients seroconverted compared with 33 (9.7%) of 341 placebo recipients, respectively (11). One U.S. study was designed specifically to evaluate the antibody responses to vaccines (DTaP-HepB-IPV, PCV7 and Hib) coadministered with RV1. A total of 180 infants received the 2 doses of RV1 coadministered with the other vaccines, and 137 infants who received the 2 RV1 doses 1 month after the other vaccines were included in the ATP cohort. Noninferiority criteria were met for all antigens, indicating that coadministration of RV1 with routine childhood vaccines did not diminish the immune responses to any of these vaccine antigens (11,108). EfficacyThe efficacy of the licensed formulation of RV1 has been evaluated in two large phase III trials among healthy infants, one conducted in 11 Latin American countries (109) and one conducted in six European countries (110) (Table 1). OPV was not coadministered; other routine childhood vaccines could be administered concomitantly. In both studies, both breast and formula feeding were permitted. In the Latin American trial, 17,867 infants enrolled into the safety study also were part of the efficacy analysis and were included in the per-protocol efficacy analysis (Table 1) (109). The primary efficacy endpoint in this study was prevention of severe wild-type rotavirus gastroenteritis from 2 weeks after second dose until age 1 year. Wild-type rotavirus gastroenteritis was defined as an episode of gastroenteritis in which rotavirus other than vaccine strain was identified in a stool sample collected no later than 7 days after symptom onset. A clinical definition for severe rotavirus gastroenteritis was used: diarrhea (three or more loose or watery stools within 24 hours), with or without vomiting, in which rotavirus other than vaccine strain was identified in a stool sample and that required overnight hospitalization or rehydration equivalent to WHO plan B (oral rehydration) or plan C (intravenous rehydration) in a medical facility. Stools were tested for the presence of rotavirus antigen by enzyme-linked immunosorbent assay (ELISA). Stools that tested positive by ELISA were analyzed further for G and P type determination by RT-PCR, followed by reverse hybridization assay or optional sequencing (108). For certain outcomes, severe rotavirus gastroenteritis also was defined as a score of >11 on an established 20-point severity scoring system (Vesikari scale) on the basis of the intensity and duration of symptoms of fever, vomiting, diarrhea, degree of dehydration, and treatment needed (109). In the Latin American trial, the efficacy of RV1 against severe rotavirus gastroenteritis (clinical definition) after completion of a 2-dose series until age 1 year was 84.7% (CI = 71.7-92.4) (109) (Table 2); the efficacy results were similar when severe rotavirus gastroenteritis was defined as an episode of rotavirus gastroenteritis with a Vesikari score of >11 (84.8%; CI = 71.1-92.7). The efficacy against severe rotavirus gastroenteritis (clinical definition) after completion of a 2-dose series until age 2 years was 80.5% (CI = 71.3-87.1). Efficacy against non-G1 strains was observed; few cases from certain strains were detected (Table 3). The efficacy against G2 was greater than zero for subjects followed to age 1 year and those followed to age 2 years, but the 95% CIs included zero. The efficacy against rotavirus gastroenteritis of any severity was not measured in the Latin American trial. For the first year follow-up period, the efficacy for 2 doses of RV1 against severe gastroenteritis (clinical definition) from any cause was 40.0% (CI = 27.7-50.4) (109). In the European trial, efficacy was assessed among 3,874 infants who received either RV1 or placebo (110). The primary efficacy endpoint in this study was prevention of wild-type rotavirus gastroenteritis of any grade of severity occurring from 2 weeks after dose 2 until the end of the first rotavirus season. In general, efficacy results were somewhat higher in the European trial than in the Latin American trial (Tables 2 and 3). The efficacy against rotavirus gastroenteritis of any severity after the 2-dose regimen until the end of the first rotavirus season was 87.1% (CI = 79.6-92.1), and efficacy against severe rotavirus gastroenteritis (score of >11 on the Vesikari scale) was 95.8% (CI = 89.6-98.7) (Table 2). The efficacy after 2 doses of RV1 through the end of the second rotavirus season was 78.9% (CI = 72.7-83.8) against rotavirus gastroenteritis of any severity, and 90.4% (CI = 85.1-94.1) against severe rotavirus gastroenteritis (score of >11 on the Vesikari scale). Efficacy against non-G1 strains was observed; few cases from certain strains were detected (Table 3). For the second season and for the combined first and second season, the efficacy against severe disease from G2 was positive with a 95% CI that did not include zero. For the first season follow-up period, the efficacy for 2 doses of RV1 against hospitalization for gastroenteritis of any cause was 74.7% (CI = 45.5-88.9). The efficacy of RV1 against rotavirus gastroenteritis of any severity through the first season among infants in the European trial that breastfed at the time of at least 1 dose (86.0%; CI = 76.8-91.9) was similar to the efficacy among infants not breastfed at the time of either dose (90.8%; CI = 72.5-97.7) (108). Efficacy against severe rotavirus gastroenteritis through the first season also was similar for the two groups (breastfed at the time of at least 1 dose: 95.7% [CI = 88.2-98.9] compared with not breastfed at the time of either dose: 96.2% [CI = 74.1-99.9]). Data on the efficacy of RV1 among preterm infants are not available. Adverse Events After VaccinationIntussusception The Latin American trial was designed as a large trial to permit evaluation of safety with respect to intussusception; 63,225 infants (including 2,060 infants from Finland) received at least 1 dose of RV1 or placebo (109). No increased risk for intussusception was observed after administration of RV1 when compared with placebo. For the prespecified period days 0-30 after either dose, on the basis of the date of diagnosis, six confirmed intussuception cases occurred among 31,673 infants who received RV1 and seven occurred among 31,552 infants who received placebo (relative risk [RR]: 0.85; CI = 0.30-2.42). On the basis of the date of intussusception onset, seven confirmed intussusception cases occurred among RV1 recipients and seven occurred among placebo recipients for the period days 0--30 after either dose (108). None of the confirmed intussusception cases in either vaccine or placebo group had onset from days 0--14 after dose 1. Other Adverse EventsDuring the entire course of eight clinical studies, 68 (0.19%) deaths occurred among 36,755 RV1 recipients, and 50 (0.15%) deaths occurred among 34,454 placebo recipients (11). The most commonly reported cause of death after vaccination was pneumonia, which occurred in 19 (0.05%) RV1 recipients and 10 (0.03%) placebo recipients (RR: 1.7; CI = 0.8-4.2). Infants were monitored for SAEs that occurred in the 31-day period after vaccination in eight clinical studies (11). Severe disease from one or more SAE occurred in 627 (1.7%) of 36,755 RV1 recipients compared with 659 (1.9%) of 34,454 placebo recipients (RR: 0.9; CI = 0.8-1.0). Diarrhea (RV1: 0.02%; placebo: 0.07%), dehydration (RV1: 0.02%; placebo: 0.06%), and gastroenteritis (RV1: 0.2%; placebo: 0.3%) occurred at a statistically higher (CI for relative risk excluded 1.0) incidence among placebo recipients compared with RV1 recipients. SAEs were coded with Medical Dictionary for Regulatory Activities (MedDRA) terms on the basis of information collected by study investigators from parental reports or medical records. Rates of SAEs were similar or the same between RV1 and placebo recipients for SAEs coded with the preferred MedDRA term "pneumonia" (RV1: 0.3%; placebo: 0.4%) and "convulsions" (RV1: 0.02%; placebo: 0.02%) (108). In the Latin American trial, no notable differences were observed in the vaccinated versus placebo groups in rates of nonfatal pneumonia events and pneumonia hospitalizations (108). However, an increase was observed in pneumonia deaths (using combined pneumonia-related preferred terms) during the period between dose 1 and visit 3 [visit 3 took place 30-90 days after dose 2]; 16 (0.05%) such deaths occurred among RV1 recipients, and six (0.02%) occurred among placebo recipients (risk difference: 3.2 per 10,000 infants; exact p = 0.035) (108). In the European trial, no deaths were reported (108); rates of SAEs with the preferred term "pneumonia" reported from dose 1 to the end of the second rotavirus season were significantly greater among RV1 recipients than among placebo recipients (0.9% and 0.3%, respectively) (risk difference: 61 per 10,000 infants; p = 0.03). In the RV1 group, 71% of the pneumonia SAEs occurred >153 days from the last dose of RV1 (111) (GSK, unpublished data, 2008). In all the other clinical trials in the BLA, and in the core integrated safety summary, statistically significant differences were not noted in the vaccine versus placebo groups for pneumonia or other pneumonia-related SAEs within the 31-day postvaccination period or for the full study period (111) (GSK, unpublished data, 2008). Excluding the Latin American safety and efficacy trial, for all other BLA trials combined, no statistically significant differences were noted among the vaccine versus placebo groups in pneumonia-related deaths during the full study period. The significance of these pneumonia-related findings is unclear. Additional data are expected from studies nearing completion in Asia and Africa (Leonard Friedland, GSK, personal correspondence, June 2008). In the Latin American trial, statistically significantly more events coded with the preferred term "convulsions" were reported from dose 1 to visit 3 in RV1 recipients (16 [0.05%]) compared with placebo recipients (6 [0.02%]; p = 0.03) (108). When convulsion-related preferred terms were combined, no statistically significant difference in these events occurred in RV1 recipients compared with placebo recipients in three periods that were analyzed: from dose 1 to visit 3 (RV1: 20 [0.06%]; placebo: 12 [0.04%]), within 31 days after any dose (RV1: seven [0.02%]; placebo: nine [0.03%]), and 43 days after any dose (RV1: 12 [0.04%]; placebo: nine [0.03%]). In the European trial, no statistically significant difference was observed between convulsion-related SAEs in the RV1 group compared with the placebo group within 31 or 43 days after any dose (one event in each group; 0.04% and 0.07%, respectively) (108). In seven clinical studies, detailed safety information for solicited adverse events was collected by parents and guardians for the day of vaccination and the next 7 days. Adverse events among RV1 recipients and placebo recipients occurred at similar rates, with the exception of Grade 3 (i.e., those that prevented normal everyday activities) cough or runny nose, which was slightly but statistically significantly higher in the RV1 group (108) (Table 6). During the 31-day period after vaccination, the following unsolicited adverse events occurred at a statistically higher incidence among RV1 recipients compared with placebo recipients: irritability (11.4% in RV1 group compared with 8.7% in the placebo group) and flatulence (2.2% in RV1 group compared with 1.3% in the placebo group) (11). No significant differences in Grade 3 irritability and flatulence were observed between the vaccine recipients and placebo recipients (108). In the placebo-controlled trials (including some that were not 1:1 randomized), Kawasaki disease was reported in 17 (0.03%) RV1 recipients and nine (0.02%) placebo recipients (RR: 1.7; CI = 0.7-4.4); one case occurred within 30 days after study dose in RV1 recipients and one in the placebo recipients (RR: 1.0; CI = 0.01-78.4) (11). Among RV1 recipients, the time of onset after study dose varied (range: 3 days--19 months). Preterm InfantsA limited number of preterm infants (reported gestational age of <36 weeks) who received RV1 were followed for serious adverse events up to 30-90 days after dose 2. Serious adverse events were observed in seven (5.2%) of 134 preterm RV1 recipients compared with six (5.0%) of 120 preterm placebo recipients (11). No deaths or cases of intussusception were reported among these infants. Additional data are expected in the near future. Shedding and Transmission of Vaccine VirusRotavirus antigen shedding in stools postvaccination was evaluated in all or a subset of infants from seven phase II or III studies in various countries (RV1 administered at 106.5--106.8 CCID50 per dose, with 26-152 infants evaluated per study) (108). After dose 1, rotavirus antigen shedding was detected by ELISA in 50.0%-80.0% (depending on study) of infants at approximately day 7, 19.2%-64.1% at approximately day 15, 0-24.3% at approximately day 30, and 0-2.6% at approximately day 60. After dose 2, rotavirus antigen shedding was detected in 4.2%-18.4% (depending on study) of infants at approximately day 7, 0-16.2% at approximately day 15, 0-1.2% at approximately day 30, and 0 at approximately day 45 (day 45 was assessed in only one study). Shedding of live rotavirus was assessed in two BLA studies in which RV1 was administered at 106.5 CCID50 per dose (108). In both studies, stool samples that were collected from a subset of infants at approximately day 7 after dose 1 were tested by ELISA. Stools that were rotavirus-antigen positive were tested subsequently for live virus by focus forming unit assay if enough sample was available. Live virus was detected in six (46.2%) of 13 and 15 (45.5%) of 33 rotavirus-antigen positive stools, for an estimated 26% of vaccinated infants shedding live virus at approximately day 7 after dose 1. The potential for transmission of vaccine virus to other persons was not assessed. Cost-Effectiveness of Rotavirus VaccinationIn a 2006 analysis that considered rotavirus disease burden, vaccine efficacy, vaccine coverage rates, and health costs, investigators estimated that a national rotavirus vaccination program in which 3 doses of RV5 were administered at ages 2, 4, and 6 months would result in 255,000 fewer physician visits, 137,000 fewer ED visits, 44,000 fewer hospitalizations, and 13 fewer deaths among children in one U.S. birth cohort followed to age 5 years (5). From the health-care perspective (i.e., evaluating medical costs only), the vaccination program was estimated to be cost-saving if the total cost per child (including administration costs) was less than $66 (in 2004 dollars) for a complete series and would incur a net cost at $143 per child. From the societal perspective (i.e., evaluating medical and nonmedical costs), vaccination was likely to be cost-saving at a total cost per child of less than $156 and would be a net cost to society if total cost of vaccination was more than $238 per child. At the manufacturer's price of $62.50 (in 2006 dollars) per dose, a rotavirus vaccination program with RV5 would cost an estimated $197,190 per life-year saved and $138 per case averted from the societal perspective. This analysis was repeated in 2008 for RV1 administered at ages 2 and 4 months (112). A national program with either the 3-dose RV5 series or the 2-dose RV1 series will have similar cost-effectiveness estimates. Assuming a total cost of $208 per child for RV1 and $218 per child for RV5 (in 2006 dollars; one extra $10 administration cost for RV5), RV1 was slightly more cost-effective than RV5 (e.g., from a societal perspective, median estimates of $94 compared with $139 per case averted and $128,400 compared with $198,546 per life-year saved, respectively). However, because of uncertainty in cost per dose, administration, and shipping for each product and of the field vaccine effectiveness of a product's full or partial series, these differences in median estimates between the vaccines might not translate into a true difference for a program. Rationale for Rotavirus Vaccination and Development of Updated RecommendationsThe rationale for adopting vaccination of infants as the primary public health measure for prevention of rotavirus disease, especially severe rotavirus disease, in the United States is threefold. First, rates of rotavirus illness among children in industrialized and less developed countries were similar, indicating that clean water supplies and good hygiene have little effect on virus transmission; therefore, further improvements in hygiene in the United States were unlikely to have a substantial impact on disease prevention (36,75,113--116). Second, in the United States, a high level of rotavirus morbidity continued in the prevaccine era despite available therapies. For example, the rate of hospitalizations for gastroenteritis in young children declined only modestly during 1979-1995 (8,117) despite the widespread availability of oral rehydration solutions in the treatment of dehydrating gastroenteritis (118,119). Third, studies of natural rotavirus infection indicated that initial infection protects against subsequent severe gastroenteritis, although subsequent asymptomatic infections and mild disease still might occur (75,76,120). Therefore, vaccination early in life, which mimics a child's first natural infection, will not prevent all subsequent disease but should prevent the majority of cases of severe rotavirus disease and their sequelae (e.g., dehydration, physician visits, hospitalizations, and deaths). In drafting and updating rotavirus vaccine recommendations for consideration by ACIP, the rotavirus vaccine work group acknowledged that differences existed in the design of the vaccine trials and studies and that these differences and the lack of a head-to-head trial between the two licensed vaccines limited direct comparisons of some study results. One aspect that differed in the trials was the maximum ages for doses of vaccine. The maximum age for dose 1 in the trial protocols differed by approximately 3 weeks (Table 1). In addition, because the RV1 series has only 2 doses of vaccine whereas the RV5 series has 3 doses, the maximum age for the last dose for the RV1 trials was younger than that for the RV5 trial. When developing the recommendations for the maximum ages for doses, the workgroup considered the vaccines' safety and efficacy data and also the effect that having the same or different maximum ages for the products would have on the ability of practitioners to follow the recommendations. After reviewing the options, the workgroup considered that harmonization of the maximum ages for doses of the two vaccines, as presented in the recommendations, would be unlikely to affect the safety and efficacy of the vaccines and would be programmatically advantageous. Changes to Recommendations from the 2006 ACIP Statement
Recommendations for the Use of Rotavirus VaccineRoutine Administration ACIP recommends routine vaccination of U.S. infants with rotavirus vaccine (Table 7). Two different rotavirus vaccine products are licensed for use in infants in the United States, RV5 and RV1. The products differ in composition and schedule of administration. Safety and efficacy were demonstrated for both vaccines in prelicensure clinical trials. Efficacy studies demonstrated that rotavirus vaccine was 85%-98% protective against severe rotavirus disease and 74%-87% protective against rotavirus disease of any severity through approximately the first rotavirus season (93,109,110). ACIP does not express a preference for either RV5 or RV1. RV5 is to be administered orally in a 3-dose series, with doses administered at ages 2, 4, and 6 months. RV1 is to be administered orally in a 2-dose series, with doses administered at ages 2 and 4 months (Table 8). The minimum age for dose 1 of rotavirus vaccine is 6 weeks; the maximum age for dose 1 is 14 weeks and 6 days. Vaccination should not be initiated for infants aged 15 weeks and 0 days or older because of insufficient data on safety of dose 1 of rotavirus vaccine in older infants. The minimum interval between doses of rotavirus vaccine is 4 weeks; no maximum interval is set. All doses should be administered by age 8 months and 0 days. For infants to whom dose 1 of rotavirus vaccine is administered inadvertently at age 15 weeks and 0 days or older, the rest of the rotavirus vaccination series should be completed according to the schedule and by age 8 months and 0 days because timing of dose 1 should not affect the safety and efficacy of any subsequent dose(s). Infants who have had rotavirus gastroenteritis before receiving the full series of rotavirus vaccination should still start or complete the schedule according to the age and interval recommendations because the initial rotavirus infection might provide only partial protection against subsequent rotavirus disease. No restrictions are placed on the infant's feeding before or after receipt of rotavirus vaccine. Breastfed infants should be vaccinated according to the same schedule as nonbreastfed infants. The efficacy of the rotavirus vaccine series is similar among breastfed and nonbreastfed infants. As with all other vaccines, rotavirus vaccine can be administered to infants with minor acute illness (e.g., mild gastroenteritis or mild upper-respiratory tract infection, with or without fever). Simultaneous AdministrationRotavirus vaccine can be administered together with DTaP vaccine, Hib vaccine, IPV, hepatitis B vaccine, and pneumococcal conjugate vaccine. Available evidence suggests that rotavirus vaccine does not interfere with the immune response to these vaccines (for each rotavirus vaccine, see Immunogenicity). The infant's immune response to influenza vaccine administered at the same time as rotavirus vaccine has not been studied. However, ACIP has recommended previously that an inactivated vaccine (e.g., inactivated influenza vaccine) may be administered either simultaneously or at any time before or after a different inactivated vaccine or live vaccine (e.g., rotavirus vaccine) (121). Interchangeability of Rotavirus VaccinesACIP recommends that the rotavirus vaccine series be completed with the same product whenever possible. However, vaccination should not be deferred because the product used for a previous dose(s) is not available or is unknown. In these situations, the provider should continue or complete the series with the product available. If any dose in the series was RV5 or the vaccine product is unknown for any dose in the series, a total of 3 doses of rotavirus vaccine should be administered. All doses should be administered by age 8 months and 0 days. No studies address the interchangeability of the two rotavirus vaccine products. However, no theoretic reason exists to expect that the risk for adverse events would be increased if the series included more than one product, compared with the risk for adverse events of a series containing only one product. Further, although it is possible that effectiveness of a series that contained both products could be reduced compared with a complete series with one product, the effectiveness of a series that contains both products is likely to be greater than an incomplete series with one product. ContraindicationsRotavirus vaccine should not be administered to infants who have a history of a severe allergic reaction (e.g., anaphylaxis) after a previous dose of rotavirus vaccine or to a vaccine component. Latex rubber is contained in the RV1 oral applicator, so infants with a severe (anaphylactic) allergy to latex should not receive RV1. The RV5 dosing tube is latex-free. PrecautionsAltered Immunocompetence Practitioners should consider the potential risks and benefits of administering rotavirus vaccine to infants with known or suspected altered immunocompetence (121); consultation with an immunologist or infectious diseases specialist is advised. Children and adults who are immunocompromised because of congenital immunodeficiency, hematopoetic transplantation, or solid organ transplantation sometimes experience severe or prolonged rotavirus gastroenteritis. However, no safety or efficacy data are available for the administration of rotavirus vaccine to infants who are immunocompromised or potentially immunocompromised, including 1) infants with primary and acquired immunodeficiency states, cellular immunodeficiencies, and hypogammaglobulinemic and dysgammaglobulinemic states; 2) infants with blood dyscrasias, leukemia, lymphomas, or other malignant neoplasms affecting the bone marrow or lymphatic system; 3) infants on immunosuppressive therapy (including high-dose systemic corticosteroids); and 4) infants who are HIV-exposed or infected. However, two considerations support vaccination of HIV-exposed or infected infants: first, the HIV diagnosis might not be established in infants born to HIV-infected mothers before the age of the first rotavirus vaccine dose (only 1.5%--3% of HIV-exposed infants in the United States will be determined to be HIV-infected); and second, vaccine strains of rotavirus are considerably attenuated. Acute GastroenteritisIn usual circumstances, rotavirus vaccine should not be administered to infants with acute moderate or severe gastroenteritis until the condition improves. However, infants with mild acute gastroenteritis can be vaccinated, particularly if the delay in vaccination might be substantial and might make the infant ineligible to receive vaccine (e.g., aged >15 weeks and 0 days before the vaccine series is started). Rotavirus vaccine has not been studied among infants with concurrent acute gastroenteritis. In these infants, the immunogenicity and efficacy of rotavirus vaccine theoretically could be compromised. For example, in some instances, infants who received OPV during an episode of acute gastroenteritis had diminished poliovirus antibody responses (122). Moderate or Severe Acute IllnessAs with all other vaccines, the presence of a moderate or severe acute illness with or without fever is a precaution to administration of rotavirus vaccine. Infants with a moderate or severe acute illness should be vaccinated as soon as they have recovered from the acute phase of the illness. This precaution avoids superimposing any potential adverse effects of the vaccine on the underlying illness or mistakenly attributing a manifestation of the underlying illness to the vaccine. Vaccination should not be delayed because of the presence of mild respiratory tract illness or other mild acute illness with or without fever. Pre-existing Chronic Gastrointestinal DiseasesInfants with pre-existing gastrointestinal conditions (e.g., congenital malabsorption syndromes, Hirschsprung's disease, or short-gut syndrome) who are not undergoing immunosuppressive therapy should benefit from receiving rotavirus vaccine, and ACIP considers the benefits to outweigh the theoretic risks. However, no data are available on the safety and efficacy of rotavirus vaccine for infants with preexisting chronic gastrointestinal conditions. Previous History of IntussusceptionPractitioners should consider the potential risks and benefits of administering rotavirus vaccine to infants with a previous history of intussusception. Available data do not indicate that RV5 or RV1 are associated with intussusception. A previously licensed rotavirus vaccine that is no longer available in the United States, RRV-TV, was associated with an increased risk for intussusception. Compared with infants who have never had intussusception, infants with a history of intussusception are at higher risk for a repeat episode of intussusception. No data are available on the administration of rotavirus vaccine to infants with a history of intussusception. Infants with Spina Bifida or Bladder ExstrophyLatex rubber is contained in the RV1 oral applicator whereas the RV5 dosing tube is latex-free. Therefore, some experts prefer that infants with spina bifida or bladder exstrophy, who are at high risk for acquiring latex allergy, receive RV5 instead of RV1 to minimize latex exposure in these children. However, if RV1 is the only rotavirus vaccine available, it should be administered, because the benefit of vaccination is considered to be greater than the risk for sensitization. Special SituationsPreterm Infants (<37 Weeks' Gestation) ACIP considers the benefits of rotavirus vaccination of preterm infants (those born at <37 weeks' gestation) to outweigh the risks of adverse events. Data suggest that preterm infants are at increased risk for hospitalization from rotavirus or other viral pathogens associated with gastroenteritis during their first one to two years of life. In clinical trials, rotavirus vaccine appeared to be generally well tolerated in preterm infants, although a relatively small number of preterm infants have been evaluated (for each rotavirus vaccine, see Adverse Events After Immunization). ACIP supports vaccination of preterm infants according to the same schedule and precautions as full-term infants and under the following conditions: the infant's chronological age meets the age requirements for rotavirus vaccine (e.g., age 6 weeks--14 weeks and 6 days for dose 1), the infant is clinically stable, and the vaccine is administered at the time of discharge from the neonatal intensive care unit [NICU] or nursery, or after discharge from the NICU or nursery. Although the lower level of maternal antibody to rotavirus in very preterm infants theoretically could increase the risk for adverse reactions from rotavirus vaccine, ACIP believes the benefits of vaccinating the infant when age-eligible, clinically stable, and no longer in the hospital outweigh the theoretic risks. Vaccine strains of rotavirus are shed in stools of vaccinated infants (for each rotavirus vaccine, see Shedding and Transmission of Vaccine Virus), so if an infant were to be vaccinated with rotavirus vaccine while still needing care in the NICU or nursery, at least a theoretic risk exists for vaccine virus being transmitted to infants in the same unit who are acutely ill (moderate or severe acute illness is a precaution for vaccination) and to preterm infants who are not age-eligible for vaccine. ACIP considers that, in usual circumstances, the risk from shedding outweighs the benefit of vaccinating the infant who is age-eligible for vaccine but who will remain in the NICU or nursery after vaccination. Exposure of Immunocompromised Persons to Vaccinated InfantsInfants living in households with persons who have or are suspected of having an immunodeficiency disorder or impaired immune status can be vaccinated. Vaccine virus (attenuated rotavirus) is shed in the stools of infants after rotavirus vaccination. However, no data are available on the risk for transmission of vaccine virus to household contacts and the risk for any subsequent disease. Vaccine virus is shed more commonly and for longer periods after RV1 than after RV5 (for each rotavirus vaccine, see Shedding and Transmission of Vaccine Virus). ACIP believes that the protection of the immunocompromised household member afforded by vaccinating the infant in the household and preventing wild-type rotavirus disease outweighs the small risk for transmitting vaccine virus to the immunocompromised household member and any subsequent theoretic risk for vaccine virus-associated disease. Vaccine virus is shed during the first weeks after administration of rotavirus vaccine; handwashing after diaper changing is always recommended. Exposure of Pregnant Women to Vaccinated InfantsInfants living in households with pregnant women should be vaccinated according to the same schedule as infants in households without pregnant women. Because the majority of women of childbearing age have preexisting immunity to rotavirus, the risk for infection and any subsequent theoretic risk for disease from potential exposure to the attenuated vaccine virus is considered to be very low. Regurgitation of VaccineThe practitioner should not readminister a dose of rotavirus vaccine to an infant who regurgitates, spits out, or vomits during or after administration of vaccine. No data exist on the benefits or risks associated with readministering a dose. The infant should receive the remaining recommended doses of rotavirus vaccine following the routine schedule (with a 4-week minimum interval between doses). Hospitalization After VaccinationIf a recently vaccinated infant is hospitalized for any reason, no precautions other than standard precautions need to be taken to prevent spread of vaccine virus in the hospital setting. Infants Who Have Recently Received or Will Receive an Antibody-Containing Blood ProductRotavirus vaccine may be administered at any time before, concurrent with, or after administration of any blood product, including antibody-containing products, following the routinely recommended schedule for rotavirus vaccine among infants who are eligible for vaccination. No data are available on the immune response to rotavirus vaccine in infants who have recently received a blood product. In theory, infants who have recently received an antibody-containing blood product might have a reduced immunologic response to a dose of oral rotavirus vaccine. However, 2 or 3 doses of vaccine are administered in the full rotavirus vaccine series, and no increased risk for adverse events is expected. Reporting of Adverse EventsAny clinically significant or unexpected adverse event that occurs after administration of rotavirus vaccine should be reported to VAERS, even if a causal relation to vaccination is not certain. The National Childhood Vaccine Injury Act requires health-care providers to maintain permanent immunization records and to report to VAERS occurrences of specific adverse events that follow selected vaccines, including rotavirus vaccine (available at http://vaers.hhs.gov/reportable.htm). VAERS reporting forms and information are available electronically at http://vaers.hhs.gov or by telephone, 1-800-822-7967. Web-based reporting by providers is encouraged and is available at https://secure.vaers.org/VaersDataEntryinto.htm. Enhanced Postlicensure Surveillance for Adverse EventsMonitoring for adverse events after introduction of rotavirus vaccine into routine vaccination programs is important, particularly in light of the previous experience with RRV-TV and its association with intussusception. The monitoring after introduction of RV1 will be similar to that conducted for RV5 and will include manufacturer-sponsored phase IV studies and enhanced review of adverse events reported to VAERS. National Vaccine Injury Compensation ProgramThe National Vaccine Injury Compensation Program (VICP), established by the National Childhood Vaccine Injury Act of 1986, is a no-fault system through which persons thought to have suffered an injury or death as a result of administration of a covered vaccine can seek compensation. Persons of all ages who receive a VICP-covered vaccine are eligible to file a claim. The program relies on a vaccine injury table listing the vaccines covered by the program and the injuries, disabilities, illnesses, and conditions (including death) for which compensation can be awarded. Claimants also can prevail for conditions not listed in the table if they can prove causation. For a claimant to be eligible for compensation, claims must be filed within a specific time period after the injury. Rotavirus vaccine is covered by VICP under the general category of rotavirus vaccines in Category XI of the Vaccine Injury Table (available at http://www.hrsa.gov/vaccinecompensation/table.htm). In this category, no condition is specified for compensation. Additional information about the program is available at http://www.hrsa.gov/vaccinecompensation or by telephone, 1-800-338-2382. Areas for Study Related to Rotavirus VaccinationSurveillance of Rotavirus Gastroenteritis Rotavirus gastroenteritis is not a reportable disease in the United States, and testing for rotavirus infection is not always performed when a child seeks medical care for acute gastroenteritis. Rotavirus disease surveillance systems need to be adequately sensitive and specific to document the effectiveness of the vaccination program. Methods of surveillance for rotavirus disease at the national level include review of national hospital discharge databases for rotavirus-specific or rotavirus-compatible diagnoses, surveillance for rotavirus disease at three sites that participate in NVSN, and reports of rotavirus detection from a sentinel system of laboratories (6,7,14). At the state and local levels, surveillance efforts at sentinel hospitals or by review of hospital discharge databases can be used to monitor the impact of the vaccine program. Special studies (e.g., case-control studies and retrospective cohort studies) will be used to measure the effectiveness of rotavirus vaccine under routine use in the United States. Detection of Unusual Strains of RotavirusCDC has established a national strain surveillance system of sentinel laboratories to monitor circulating rotavirus strains before and after the introduction of rotavirus vaccine (64--66). This system is designed to detect new or unusual strains causing gastroenteritis that might not be prevented effectively by vaccination, which might affect the success of the vaccination program. ResearchAdditional studies would be valuable to evaluate the safety and efficacy of rotavirus vaccine administered to infants who are born preterm, have immune deficiencies, live in households with immunocompromised persons, have chronic gastrointestinal disease, or start the series late. Postlicensure studies also could determine the relative effectiveness of rotavirus vaccine when less than the full series is administered and evaluate possible secondary transmission of vaccine virus. Acknowledgments Assistance was provided by Edward Belongia, MD, and staff at the Vaccine Safety Datalink sites (Marshfield Clinic Research Foundation, Marshfield, Wisconsin; Health Partners Research Foundation, Minneapolis, Minnesota; Kaiser Permanente of Colorado, Denver, Colorado; Kaiser Permanente of Northern California, Oakland, California; Group Health Cooperative, Seattle Washington; Northwest Kaiser Permanente, Portland, Oregon; Harvard Pilgrim/Harvard Vanguard, Boston Massachusetts; and Southern California Kaiser Permanente, Los Angeles, California); Kathleen Neuzil, MD, PATH and the University of Washington; Rosemary Tiernan, MD, Paul Kitustani, MD, Food and Drug Administration; Penina Haber, MPH, James Baggs, PhD, Office of the Chief Science Officer, Larry Pickering, MD, Office of the Director, Haley Clayton, MPH, Lauren Stockman, MPH; Jon Gentsch, PhD, Marc-Alain Widdowson, DVM, National Center for Immunization and Respiratory Diseases; Martin Meltzer, PhD, National Center for Preparedness, Detection, and Control of Infectious Diseases, CDC. References
* In these recommendations, the term “rotavirus vaccine” is used to refer to both RV5 and RV1.
Advisory Committee on Immunization PracticesMembership List as of June 2008
ACIP Rotavirus Vaccine Working GroupChair: Lance Chilton, MD, Albuquerque, New Mexico.
![BOX. Characteristics of RotaTeq® (RV5) and Rotarix® (RV1)
Characteristic
RV5
RV1
Parent rotavirus strain
Bovine strain WC3 (type G6P7[5])
Human strain 89-12 (type G1P1A[8])
Vaccine composition
Reassortant strains
G1 x WC3; G2 x WC3; G3 x WC3; G4 x WC3; P1A[8] x WC3
Human strain 89-12 (type G1P1A[8])
Vaccine titer
≥2.0−2.8 x 106 infectious units (IU) per dose, depending on serotype
≥106.0 median cell culture infective dose (CCID50) after reconstitution, per dose
Cell culture substrate
Vero cells
Vero cells
Formulation
Liquid requiring no reconstitution
Vial of lyophilized vaccine with a prefilled oral applicator of liquid diluent (1 ml)
Applicator
Latex-free dosing tube
Tip cap and rubber plunger of the oral applicator contain dry natural latex rubber. The vial stopper and transfer adapter are latex-free.
Other content
Sucrose, sodium citrate, sodium phosphate monobasic monohydrate, sodium hydroxide, polysorbate 80, cell culture media, and trace amounts of fetal bovine serum.
Lyophilized vaccine: amino acids, dextran, Dulbecco’s Modified Eagle Medium, sorbitol, and sucrose.
Liquid diluent contains calcium carbonate, sterile water, and xanthan
Preservatives
None
None
Shelf life
24 months
24 months
Storage
Store refrigerated at 36ºF–46ºF (2ºC–8ºC). Administer as soon as possible after being removed from refrigeration. Protect from light.
Storage before reconstitution: Refrigerate vials of lyophilized vaccine at 36ºF–46ºF (2ºC–8ºC); diluent may be stored at a controlled room temperature of 68ºF–77ºF (20ºC–25ºC). Protect vials from light.
Storage after reconstitution: Administer within 24 hours of reconstitution. May be stored refrigerated at 36ºF–46ºF (2ºC–8ºC) or at room temperature up to 77ºF (25ºC), after reconstitution.
Volume per dose
2 ml
1 ml](figures/r802a1b.gif) Return to top. Table 1 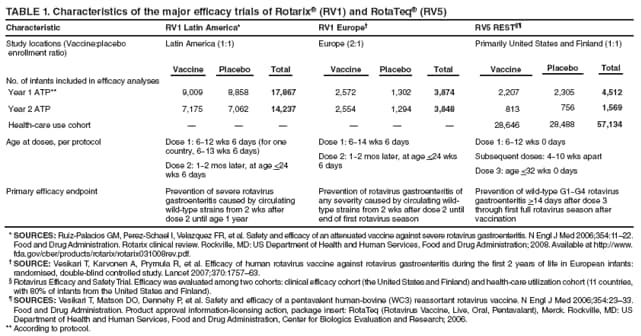 Return to top. Figure 1 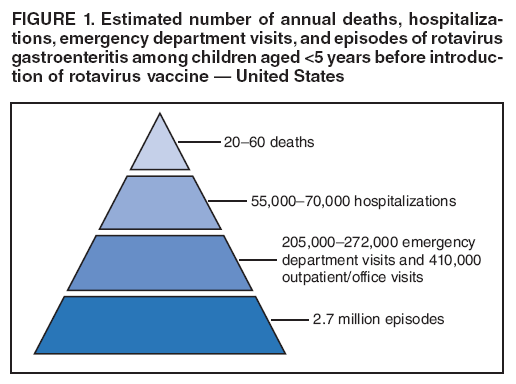 Return to top. Table 2 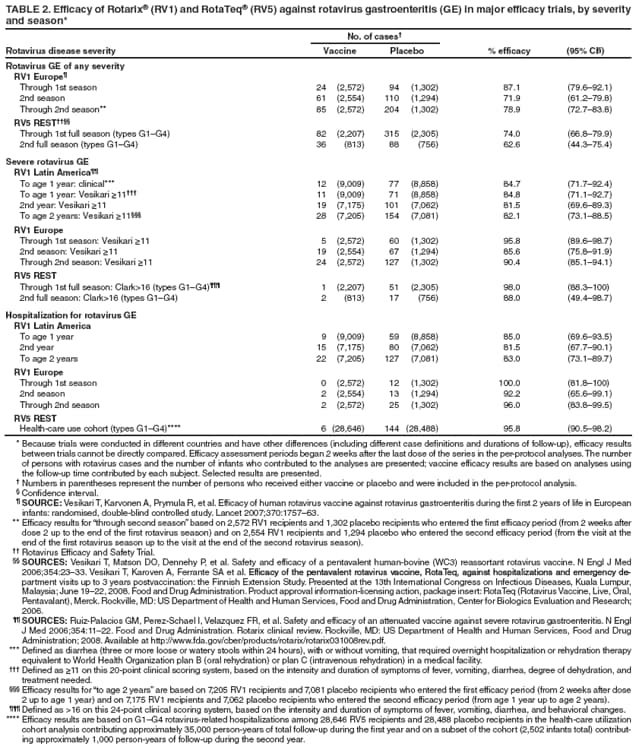 Return to top. Figure 2 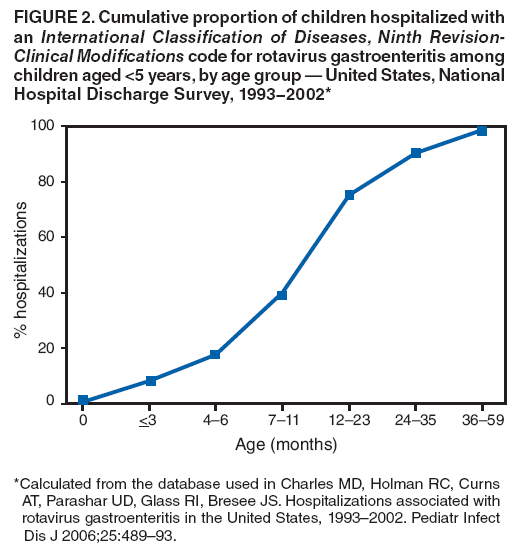 Return to top. Table 3 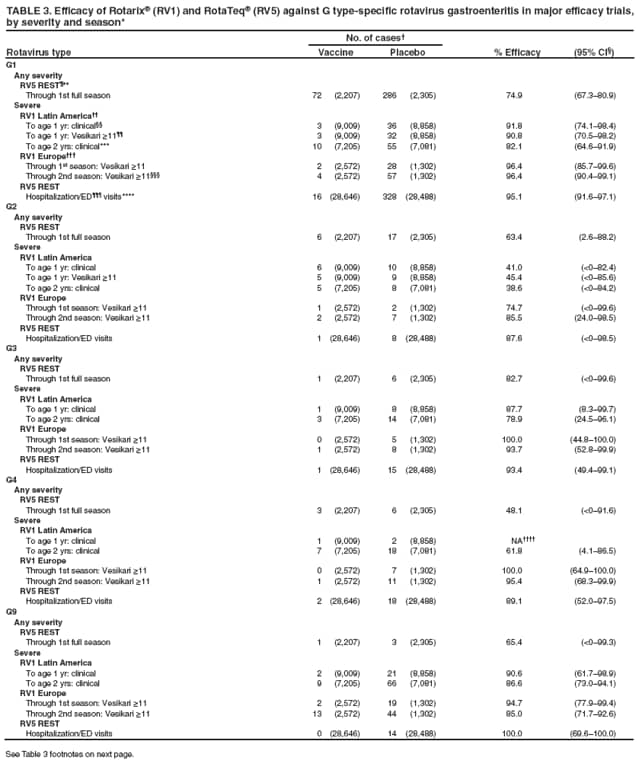  Return to top. Figure 3 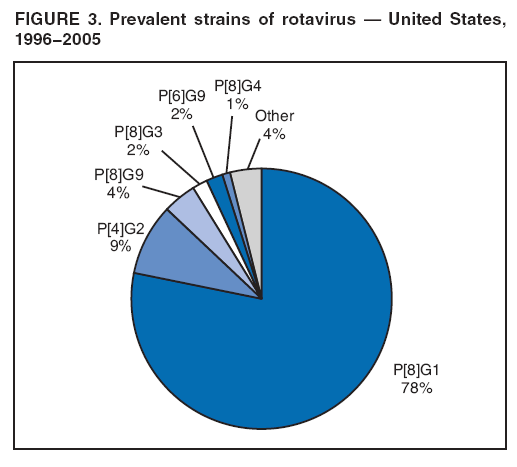 Return to top. Table 4 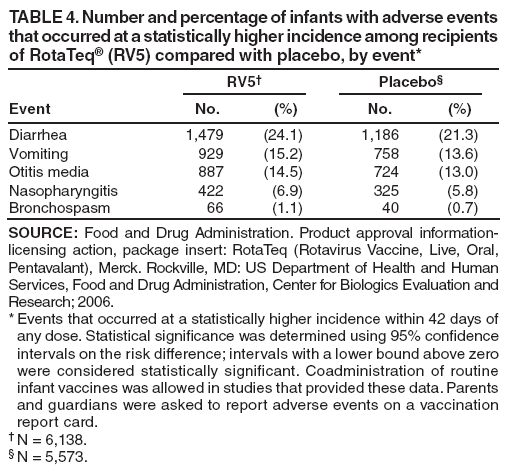 Return to top. Figure 4 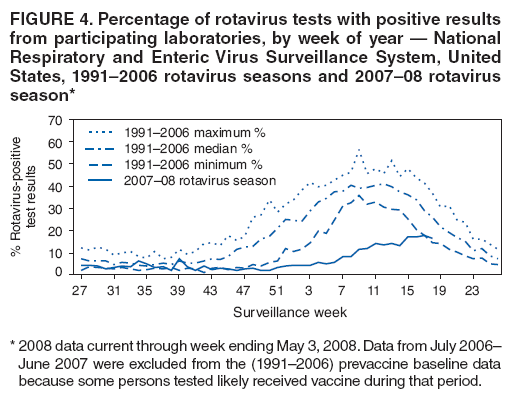 Return to top. Table 5 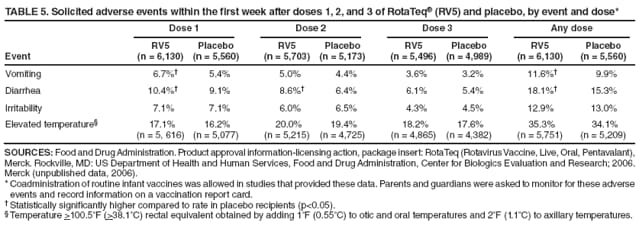 Return to top. Table 6 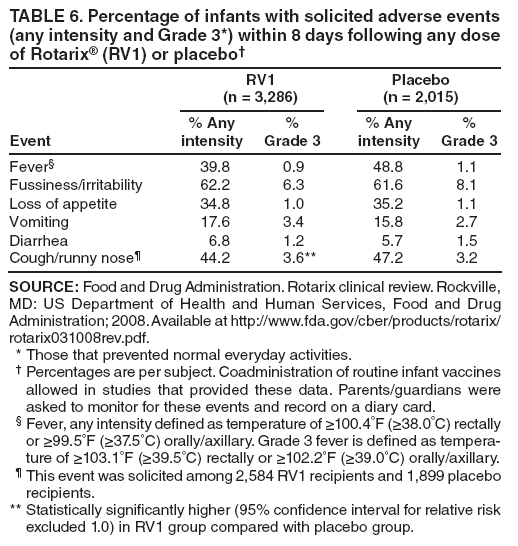 Return to top. Table 7  Return to top. Table 8 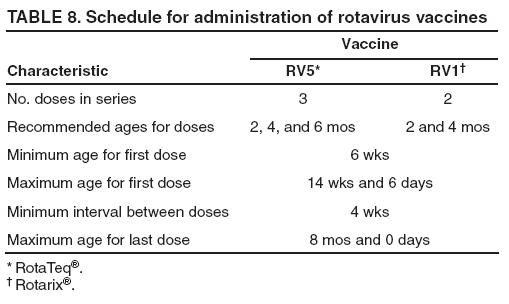 Return to top.
All MMWR HTML versions of articles are electronic conversions from typeset documents. This conversion might result in character translation or format errors in the HTML version. Users are referred to the electronic PDF version (http://www.cdc.gov/mmwr) and/or the original MMWR paper copy for printable versions of official text, figures, and tables. An original paper copy of this issue can be obtained from the Superintendent of Documents, U.S. Government Printing Office (GPO), Washington, DC 20402-9371; telephone: (202) 512-1800. Contact GPO for current prices. **Questions or messages regarding errors in formatting should be addressed to mmwrq@cdc.gov.Date last reviewed: 2/3/2009 |
|||||||||
|