 |
|
|
|
|
|
|
| ||||||||||
|
|
|
|
|
|
|
||||
| ||||||||||
|
|
|
|
|
Persons using assistive technology might not be able to fully access information in this file. For assistance, please send e-mail to: mmwrq@cdc.gov. Type 508 Accommodation and the title of the report in the subject line of e-mail. Guidelines for Using Antiretroviral Agents Among HIV-Infected Adults and AdolescentsRecommendations of the Panel on Clinical Practices for Treatment of HIV*Note: Data regarding use of hydroxyurea in combination with antiretroviral agents are limited; therefore, this report does not include recommendations regarding its use in treating persons infected with human immunodeficiency virus. Prepared by The material in this report was prepared for publication by the National Center for HIV, STD, and TB Prevention, Harold W. Jaffe, M.D., Acting Director, and the Division of HIV/AIDS Prevention — Surveillance and Epidemiology, Robert S. Janssen, M.D., Director. SummaryThe availability of an increasing number of antiretroviral agents and the rapid evolution of new information has introduced substantial complexity into treatment regimens for persons infected with human immunodeficiency virus (HIV). In 1996, the Department of Health and Human Services and the Henry J. Kaiser Family Foundation convened the Panel on Clinical Practices for the Treatment of HIV to develop guidelines for clinical management of HIV-Infected adults and adolescents (CDC. Report of the NIH Panel To Define Principles of Therapy of HIV Infection and Guidelines for the use of antiretroviral agents in HIV-infected adults and adolescents. MMWR 1998;47[RR-5]:1--41). This report, which updates the 1998 guidelines, addresses 1) using testing for plasma HIV ribonucleic acid levels (i.e., viral load) and CD4+ T cell count; 2) using testing for antiretroviral drug resistance; 3) considerations for when to initiate therapy; 4) adherence to antiretroviral therapy; 5) considerations for therapy among patients with advanced disease; 6) therapy-related adverse events; 7) interruption of therapy; 8) considerations for changing therapy and available therapeutic options; 9) treatment for acute HIV infection; 10) considerations for antiretroviral therapy among adolescents; 11) considerations for antiretroviral therapy among pregnant women; and 12) concerns related to transmission of HIV to others. Antiretroviral regimens are complex, have serious side effects, pose difficulty with adherence, and carry serious potential consequences from the development of viral resistance because of nonadherence to the drug regimen or suboptimal levels of antiretroviral agents. Patient education and involvement in therapeutic decisions is critical. Treatment should usually be offered to all patients with symptoms ascribed to HIV infection. Recommendations for offering antiretroviral therapy among asymptomatic patients require analysis of real and potential risks and benefits. Treatment should be offered to persons who have <350 CD4+ T cells/mm3 or plasma HIV ribonucleic acid (RNA) levels of >55,000 copies/mL (by b-deoxyribonucleic acid [bDNA] or reverse transcriptase-polymerase chain reaction [RT-PCR] assays). The recommendation to treat asymptomatic patients should be based on the willingness and readiness of the person to begin therapy; the degree of existing immunodeficiency as determined by the CD4+ T cell count; the risk for disease progression as determined by the CD4+ T cell count and level of plasma HIV RNA; the potential benefits and risks of initiating therapy in an asymptomatic person; and the likelihood, after counseling and education, of adherence to the prescribed treatment regimen. Treatment goals should be maximal and durable suppression of viral load, restoration and preservation of immunologic function, improvement of quality of life, and reduction of HIV-related morbidity and mortality. Results of therapy are evaluated through plasma HIV RNA levels, which are expected to indicate a 1.0 log10 decrease at 2--8 weeks and no detectable virus (<50 copies/mL) at 4--6 months after treatment initiation. Failure of therapy at 4--6 months might be ascribed to nonadherence, inadequate potency of drugs or suboptimal levels of antiretroviral agents, viral resistance, and other factors that are poorly understood. Patients whose therapy fails in spite of a high level of adherence to the regimen should have their regimen changed; this change should be guided by a thorough drug treatment history and the results of drug-resistance testing. Because of limitations in the available alternative antiretroviral regimens that have documented efficacy, optimal changes in therapy might be difficult to achieve for patients in whom the preferred regimen has failed. These decisions are further confounded by problems with adherence, toxicity, and resistance. For certain patients, participating in a clinical trial with or without access to new drugs or using a regimen that might not achieve complete suppression of viral replication might be preferable. Because concepts regarding HIV management are evolving rapidly, readers should check regularly for additional information and updates at the HIV/AIDS Treatment Information Service website (http://www.hivatis.org). IntroductionThis report was developed by the Panel on Clinical Practices for Treatment of HIV (the Panel), which was convened by the Department of Health and Human Services (DHHS) and the Henry J. Kaiser Family Foundation in 1996. The goal of these recommendations is to provide evidence-based guidance for clinicians and other health-care providers who use antiretroviral agents in treating adults and adolescents† infected with human immunodeficiency virus (HIV), including pregnant women. Although the pathogenesis of HIV infection and the general virologic and immunologic principles underlying the use of antiretroviral therapy are similar for all HIV-infected persons, unique therapeutic and management considerations exist for HIV-infected children. Therefore, guidance for antiretroviral therapy for pediatric HIV infection is not contained in this report. A separate report addresses pediatric-specific concerns related to antiretroviral therapy and is available at http://www.hivatis.org. These guidelines serve as a companion to the therapeutic principles from the National Institutes of Health (NIH) Panel To Define Principles of Therapy of HIV Infection (1). Together, the reports provide pathogenesis-based rationale for therapeutic strategies as well as guidelines for implementing these strategies. Although the guidelines represent the state of knowledge regarding the use of antiretroviral agents, this is an evolving science and the availability of new agents or new clinical data regarding the use of existing agents will change therapeutic options and preferences. Because this report needs to be updated periodically, a subgroup of the Panel on Clinical Practices for Treatment of HIV Infection, the Antiretroviral Working Group, meets monthly to review new data. Recommendations for changes are then submitted to the Panel and incorporated as appropriate.§ These recommendations are not intended to supercede the judgment of clinicians who are knowledgeable in the care of HIV-infected persons. Furthermore, the Panel recommends that, when possible, the treatment of HIV-infected patients should be directed by a clinician who has extensive experience in the care of these patients. When this is not possible, the patient should have access to such clinical experience through consultations. Each recommendation is accompanied by a rating that includes a letter and a Roman numeral (Table 1) and is similar to the rating schemes used in previous guidelines concerning prophylaxis of opportunistic infections (OIs) issued by the U.S. Public Health Service and the Infectious Diseases Society of America (2). The letter indicates the strength of the recommendation, which is based on the opinion of the Panel, and the Roman numeral reflects the nature of the evidence supporting the recommendation (Table 1). Thus, recommendations made on the basis of data from clinical trials with clinical results are differentiated from those made on the basis of laboratory results (e.g., CD4+ T lymphocyte count or plasma HIV ribonucleic acid [RNA] levels). When clinical trial data are unavailable, recommendations are made on the basis of the opinions of persons experienced in the treatment of HIV infection and familiar with the relevant literature. Testing for Plasma HIV RNA Levels and CD4+ T Cell Count To Guide Decisions Regarding TherapyDecisions regarding initiation or changes in antiretroviral therapy should be guided by monitoring the laboratory parameters of plasma HIV RNA (viral load) and CD4+ T cell count in addition to the patient's clinical condition. Results of these laboratory tests provide clinicians with key information regarding the virologic and immunologic status of the patient and the risk for disease progression to acquired immunodeficiency syndrome (AIDS) (3,4). HIV viral load testing has been approved by the Food and Drug Administration (FDA) for determining prognosis and for monitoring the response to therapy only for the reverse transcriptase-polymerase chain reaction (RT- PCR) assay and in vitro nucleic amplification test for HIV-RNA (NucliSens® HIV-1 QT, manufactured by Organon Teknika). Multiple analyses among >5,000 patients who participated in approximately 18 trials with viral load monitoring indicated a statistically significant dose-response--type association between decreases in plasma viremia and improved clinical outcome on the basis of standard results of new AIDS-defining diagnoses and survival. This relationship was observed throughout a range of patient baseline characteristics, including pretreatment plasma RNA level, CD4+ T cell count, and previous drug experience. Thus, viral load testing is an essential parameter in deciding to initiate or change antiretroviral therapies. Measurement of plasma HIV RNA levels (i.e., viral load) by using quantitative methods should be performed at the time of diagnosis and every 3--4 months thereafter for the untreated patient (AIII) (Table 2). CD4+ T cell counts should be measured at the time of diagnosis and every 3--6 months thereafter (AIII). These intervals between tests are recommendations only, and flexibility should be exercised according to the circumstances of each patient. Plasma HIV RNA levels should also be measured immediately before and again at 2--8 weeks after initiation of antiretroviral therapy (AIII). This second measurement allows the clinician to evaluate initial therapy effectiveness because, for the majority of patients, adherence to a regimen of potent antiretroviral agents should result in a substantial decrease (~1.0 log10) in viral load by 2--8 weeks. A patient's viral load should continue to decline during the following weeks and, for the majority of patients, should decrease below detectable levels (i.e., defined as <50 RNA copies/mL of plasma) by 16--24 weeks. Rates of viral load decline toward undetectable are affected by the baseline CD4+ T cell count, the initial viral load, potency of the regimen, adherence to the regimen, previous exposure to antiretroviral agents, and the presence of any OIs. These differences must be considered when monitoring the effect of therapy. However, the absence of a virologic response of the magnitude discussed previously should prompt the clinician to reassess patient adherence, rule out malabsorption, consider repeat RNA testing to document lack of response, or consider a change in drug regimen. After the patient is on therapy, HIV RNA testing should be repeated every 3--4 months to evaluate the continuing effectiveness of therapy (AII). With optimal therapy, viral levels in plasma at 24 weeks should be undetectable (5). Data from clinical trials demonstrate that lowering plasma HIV RNA to <50 copies/mL is associated with increased duration of viral suppression, compared with reducing HIV RNA to levels of 50--500 copies/mL (6). If HIV RNA remains detectable in plasma after 16--24 weeks of therapy, the plasma HIV RNA test should be repeated to confirm the result and a change in therapy should be considered (see Changing a Failing Regimen) (BIII). When deciding on therapy initiation, the CD4+ T lymphocyte count and plasma HIV RNA measurement should be performed twice to ensure accuracy and consistency of measurement (BIII). However, among patients with advanced HIV disease, antiretroviral therapy should be initiated after the first viral load measurement is obtained to prevent a potentially deleterious delay in treatment. The requirement for two measurements of viral load might place a substantial financial burden on patients or payers. Nonetheless, the Panel believes that two measurements of viral load will provide the clinician with the best information for subsequent patient follow-up. Plasma HIV RNA levels should not be measured during or within 4 weeks after successful treatment of any intercurrent infection, resolution of symptomatic illness, or immunization. Because differences exist among commercially available tests, confirmatory plasma HIV RNA levels should be measured by using the same laboratory and the same technique to ensure consistent results. A minimal change in plasma viremia is considered to be a threefold or 0.5-log10 increase or decrease. A substantial decrease in CD4+ T lymphocyte count is a decrease of >30% from baseline for absolute cell numbers and a decrease of >3% from baseline in percentages of cells (7). Discordance between trends in CD4+ T cell numbers and plasma HIV RNA levels was documented among 20% of patients in one cohort studied (8). Such discordance can complicate decisions regarding antiretroviral therapy and might be caused by factors that affect plasma HIV RNA testing. Viral load and trends in viral load are believed to be more informative for decision-making regarding antiretroviral therapy than are CD4+ T cell counts; however, exceptions to this rule do occur (see Changing a Failing Regimen). In certain situations, consultation with a specialist should be considered. Drug-Resistance TestingTesting for HIV resistance to antiretroviral drugs is a useful tool for guiding antiretroviral therapy. When combined with a detailed drug history and efforts in maximizing drug adherence, these assays might maximize the benefits of antiretroviral therapy. Studies of treatment-experienced patients have reported strong associations between the presence of drug resistance, identified by genotyping or phenotyping resistance assays, and failure of the antiretroviral treatment regimen to suppress HIV replication. Genotyping assays detect drug-resistance mutations that are present in the relevant viral genes (i.e., reverse transcriptase and protease). Certain genotyping assays involve sequencing of the entire reverse transcriptase and protease genes, whereas others use probes to detect selected mutations that are known to confer drug resistance. Genotyping assays can be performed rapidly, and results can be reported within 1--2 weeks of sample collection. Interpretation of test results requires knowledge of the mutations that are selected for by different antiretroviral drugs and of the potential for cross-resistance to other drugs conferred by certain mutations.¶ Consultation with a specialist in HIV drug resistance is encouraged and can facilitate interpretation of genotypic test results. Phenotyping assays measure a virus' ability to grow in different concentrations of antiretroviral drugs. Automated, recombinant phenotyping assays are commercially available with results available in 2--3 weeks; however, phenotyping assays are more costly to perform, compared with genotypic assays. Recombinant phenotyping assays involve insertion of the reverse transcriptase and protease gene sequences derived from patient plasma HIV RNA into the backbone of a laboratory clone of HIV either by cloning or in vitro recombination. Replication of the recombinant virus at different drug concentrations is monitored by expression of a reporter gene and is compared with replication of a reference HIV strain. Drug concentrations that inhibit 50% and 90% of viral replication (i.e., the median inhibitory concentration [IC] IC50 and IC90) are calculated, and the ratio of the IC50 of the test and reference viruses is reported as the fold increase in IC50 (i.e., fold resistance). Interpretation of phenotyping assay results is complicated by the paucity of data regarding the specific resistance level (i.e., fold increase in IC50) that is associated with drug failure; again, consultation with a specialist can be helpful for interpreting test results. Further limitations of both genotyping and phenotyping assays include the lack of uniform quality assurance for all available assays, relatively high cost, and insensitivity for minor viral species. If drug-resistant viruses are present but constitute <10%--20% of the circulating virus population, they probably will not be detected by available assays. This limitation is critical when interpreting data regarding susceptibility to drugs that the patient has taken in the past but that are not part of the current antiretroviral regimen. If drug resistance had developed to a drug that was subsequently discontinued, the drug-resistant virus can become a minor species because its growth advantage is lost (9). Consequently, resistance assays should be performed while the patient is taking his or her antiretroviral regimen, and data substantiating the absence of resistance should be interpreted cautiously in relation to the previous treatment history. Using Resistance Assays in Clinical PracticeResistance assays can be useful for patients experiencing virologic failure while on antiretroviral therapy and patients with acute HIV infection (Table 3). Recent prospective data supporting drug-resistance testing in clinical practice are derived from trials in which the test utility was assessed for cases of virologic failure. Two studies compared virologic responses to antiretroviral treatment regimens when genotyping resistance tests were available to guide therapy (10,11) with the responses observed when changes in therapy were guided by clinical judgment only. The results of both studies indicated that the short-term virologic response to therapy was substantially increased when results of resistance testing were available. Similarly, a prospective, randomized, multicenter trial demonstrated that therapy selected on the basis of phenotypic resistance testing substantially improves the virologic response to antiretroviral therapy, compared with therapy selected without the aid of phenotypic testing (12). Thus, resistance testing appears to be a useful tool in selecting active drugs when changing antiretroviral regimens in cases of virologic failure (BII). Similar rationale applies to the potential use of resistance testing for patients with suboptimal viral load reduction (see Criteria for Changing Therapy) (BIII). Virologic failure regarding highly active antiretroviral therapy (HAART) is, for certain patients, associated with resistance to one component of the regimen only (13); in that situation, substituting individual drugs in a failing regimen might be possible, although this concept requires clinical validation (see Changing a Failing Regimen). No prospective data exist to support using one type of resistance assay over another (i.e., genotyping versus phenotyping) in different clinical situations. Therefore, one type of assay is recommended per sample; however, for patients with a complex treatment history, both assays might provide critical and complementary information. Transmission of drug-resistant HIV strains has been documented and might be associated with a suboptimal virologic response to initial antiretroviral therapy (14--17). If the decision is made to initiate therapy in a person with acute HIV infection, using resistance testing to optimize the initial antiretroviral regimen is a reasonable, albeit untested, strategy (18--19) (CIII). Because of its more rapid turnaround time, using a genotypic assay might be preferred in this situation. Using resistance testing before initiation of antiretroviral therapy among patients with chronic HIV infection is not recommended (DIII) because of uncertainty regarding the prevalence of resistance among treatment-naïve persons. In addition, available resistance assays might fail to detect drug-resistant species that were transmitted when primary infection occurred but became a minor species in the absence of selective drug pressure. Reserving resistance testing for patients with suboptimal viral load suppression after therapy initiation is preferable, although this approach might change as additional information becomes available related to the prevalence of resistant virus among antiretroviral-naïve patients. Recommendations for resistance testing during pregnancy are the same as for nonpregnant women; acute HIV infection, virologic failure while on an antiretroviral regimen, or suboptimal viral load suppression after initiation of antiretroviral therapy are all appropriate indications for resistance testing. If an HIV-positive pregnant woman is taking an antiretroviral regimen that does not include zidovudine, or if zidovudine was discontinued because of maternal drug resistance, intrapartum and neonatal zidovudine prophylaxis should be administered to prevent mother-to-child HIV transmission (see Considerations for Antiretroviral Therapy Among HIV-Infected Pregnant Women). Not all of zidovudine's activity in preventing mother-to-child HIV transmission can be accounted for by its effect on maternal viral load (20); furthermore, preliminary data indicate that the rate of perinatal transmission after zidovudine prophylaxis might not differ between those with and without zidovudine-resistance mutations (21,22). Studies are needed to determine the best strategy to prevent mother-to-child HIV transmission in the presence of zidovudine resistance. Considerations for Patients with Established HIV InfectionPatients with established HIV infection are discussed in two arbitrarily defined clinical categories: asymptomatic infection or symptomatic disease (i.e., wasting, thrush, or unexplained fever for >2 weeks) including AIDS, as classified by CDC in 1993 (23). All patients in the second category should be offered antiretroviral therapy. Initiating antiretroviral therapy among patients in the first category is complex and, therefore, discussed separately. However, before initiating therapy for any patient, the following evaluation should be performed:
Additional evaluation should include routine tests relevant to preventing OIs, if not already performed (e.g., rapid plasma reagin or Venereal Disease Research Laboratory test; tuberculin skin test; toxoplasma immunoglobulin G serology; hepatitis B and C serology; and gynecologic exam, including Papanicolaou smear). Other tests are recommended, if clinically indicated (e.g., chest radiograph and ophthalmologic exam) (AII). Cytomegalovirus serology can be useful for certain patients (2) (BIII). Considerations for Initiating Therapy for the Patient with Asymptomatic HIV InfectionAlthough randomized clinical trials provide strong evidence for treating patients with <200 CD4+ T cells/mm3 (24--26), the optimal time to initiate antiretroviral therapy among asymptomatic patients with CD4+ T cell counts >200 cells/mm3 is unknown. For persons with >200 CD4+ T cells/mm3, the strength of the recommendation for therapy must balance the readiness of the patient for treatment, consideration of the prognosis for disease-free survival as determined by baseline CD4+ T cell count and viral load levels, and assessment of the risks and potential benefits associated with initiating antiretroviral therapy. Regarding a prognosis that is based on the patient's CD4+ T cell count and viral load, data are absent concerning clinical endpoints from randomized, controlled trials for persons with >200 CD4+ T cells/mm3 to guide decisions on when to initiate therapy. However, despite their limitations, observational cohorts of HIV-infected persons either treated or untreated with antiretroviral therapy provide key data to assist in risk assessment for disease progression. Observational cohorts have provided critical data regarding the prognostic influence of viral load and CD4+ T cell count in the absence of treatment. These data indicate a strong relationship between plasma HIV RNA levels and CD4+ T cell counts in terms of risk for progression to AIDS for untreated persons and provide potent support for the conclusion that therapy should be initiated before the CD4+ T cell count declines to <200 cells/mm3 (Figure; Tables 4,5). In addition, these studies are useful for the identification of asymptomatic persons at high risk who have CD4+ T cell counts >200 cells/mm3 and who might be candidates for antiretroviral therapy or more frequent CD4+ T cell count monitoring. Regarding CD4+ T cell count monitoring, the Multicenter AIDS Cohort Study (MACS) demonstrated that the 3-year risk for progression to AIDS was 38.5% among patients with 201--350 CD4+ T cells/mm3, compared with 14.3% for patients with CD4+ T cell counts >350 cells/mm3. However, the short-term risk for progression also was related to the level of plasma HIV RNA, and the risk was relatively low for those persons with <20,000 copies/mL. An evaluation of 231 persons with CD4+ T cell counts of 201--350 cells/mm3 demonstrated that the 3-year risk for progression to AIDS was 4.1% for the 74 patients with HIV RNA <20,000; 36.4% for those 53 patients with HIV RNA 20,001--55,000 copies/mL; and 64.4% for those 104 patients with HIV RNA >55,000 copies/mL. Similar risk gradations by viral load were evident for patients with CD4+ T cell counts >350 cells/mm3 (Figure; Table 5) (unpublished data, Alvaro Muñoz, Ph.D., Johns Hopkins University, Baltimore, Maryland, 2001). These data indicate that for certain patients with CD4+ T cell counts >200 cells/mm3, the 3-year risk for disease progression to AIDS in the absence of treatment is substantially increased. Thus, although observational studies of untreated persons cannot assess the effects of therapy and, therefore, cannot determine the optimal time to initiate therapy, these studies do provide key guidance regarding the risks for progression in the absence of therapy on the basis of a patient's CD4+ T cell count and viral load. Data from observational studies of HAART-treated cohorts also provide critical information to guide using antiretroviral therapy among asymptomatic patients (27--30). A collaborative analysis of data from 13 cohort studies from Europe and North America demonstrates that among drug-naïve patients without AIDS-defining illness and a viral load of <100,000 copies/mL, the 3-year probability of progression to AIDS or death was 15.8% among those who initiated therapy with CD4+ T cell counts of 0--49 cells/mm3; 12.5% among those with CD4+ T cell counts of 50--99 cells/mm3; 9.3% among those with CD4+ T cell counts of 100--199 cells/mm3; 4.7% among those with CD4+ T cell counts of 200--349 cells/mm3; and 3.4% among those with CD4+ T ell counts of 350 cells/mm3 or higher (30). These data indicate that the prognosis might be better for patients who initiate therapy at >200 cells/mm3; but risk after initiation of therapy does not vary considerably at >200 cells/mm3. However, risk for progression also was related to plasma HIV RNA levels in this study. A substantial increase in risk for progression was evident among all patients with a viral load >100,000 copies/mL. In other cohort studies, an apparent benefit in terms of disease progression was reported among persons who began antiretroviral therapy when CD4+ T cell counts were >350 cells/mm3, compared with those who deferred therapy (31,32). For example, in the Swiss cohort study, an approximate 7-fold decrease occurred in disease progression to AIDS among persons who initiated therapy with a CD4+ T cell count >350 cells/mm3, compared with those who were monitored without therapy during a 2-year period (32). However, a substantial incidence of adverse treatment effects occurred among patients who initiated therapy; 40% of patients had >1 treatment changes because of adverse effects, and 20% were no longer receiving treatment after 2 years (32). Unfortunately, observational studies of persons treated with HAART also have limitations regarding the ability to determine an optimal time to initiate therapy. The relative risks for disease progression for persons with CD4+ T cell counts of 200--349 and >350 cells/mm3 cannot be precisely compared because of the low level of disease progression among these patients during the follow-up period. In addition, groups might differ in key known and unknown prognostic factors that bias the comparison. In addition to the risks for disease progression, the decision to initiate antiretroviral therapy also is influenced by an assessment of other potential risks and benefits associated with treatment. Potential benefits and risks of early or delayed therapy initiation for the asymptomatic patient should be considered by the clinician and patient (Table 5). Potential benefits of early therapy include 1) earlier suppression of viral replication; 2) preservation of immune function; 3) prolongation of disease-free survival; and 4) decrease in the risk for viral transmission. Risks include 1) the adverse effects of the drugs on quality of life; 2) the inconvenience of the majority of the available suppressive regimens, leading to reduced adherence; 3) development of drug resistance because of suboptimal suppression of viral replication; 4) limitation of future treatment options as a result of premature cycling of the patient through the available drugs; 5) the risk for transmission of virus resistant to antiretroviral drugs; 6) serious toxicities associated with certain antiretroviral drugs (e.g., elevations in serum levels of cholesterol and triglycerides, alterations in the distribution of body fat, or insulin resistance and diabetes mellitus); and 7) the unknown durability of effect of available therapies. Potential benefits of delayed therapy include 1) minimization of treatment-related negative effects on quality of life and drug-related toxicities; 2) preservation of treatment options; and 3) delay in the development of drug resistance. Potential risks of delayed therapy include 1) the possibility that damage to the immune system, which might otherwise be salvaged by earlier therapy, is irreversible; 2) the possibility that suppression of viral replication might be more difficult at a later stage of disease; and 3) the increased risk for HIV transmission to others during a longer untreated period. Finally, for certain persons, ascertaining the precise time at which the CD4+ T cell count will decrease to a level where the risk for disease is high might be difficult, and time might be required to identify an effective, tolerable regimen. This task might be better accomplished before reaching a CD4+ T cell count of 200 cells/mm3. After considering available data in terms of the relative risk for progression to AIDS at certain CD4+ T cell counts and viral loads and the potential risks and benefits associated with initiating therapy, certain specialists in this area believe that the evidence supports initiating therapy for asymptomatic HIV-infected persons with a CD4+ T cell count of <350 cells/mm3 or a viral load of >55,000 copies/mL (by RT-PCR or b-deoxyribonucleic acid [bDNA] assays). For asymptomatic patients with CD4+ T cell counts >350 cells/mm3, rationale exists for both conservative and aggressive approaches to therapy. The conservative approach is based on the recognition that robust immune reconstitution still occurs in the majority of patients who initiate therapy with CD4+ T cell counts in the 200--350 cells/mm3 range, and that toxicities and adherence challenges might outweigh benefits of initiating therapy at CD4+ T cell counts >350 cells/mm3. In the conservative approach, increased levels of plasma HIV RNA (i.e., >55,000 by RT-PCR or bDNA assays) are an indication that more frequent monitoring of CD4+ T cell counts and plasma HIV RNA levels is needed, but not necessarily for initiation of therapy. In the aggressive approach, asymptomatic patients with CD4+ T cell counts >350 cells/mm3 and levels of plasma HIV RNA >55,000 copies/mL would be treated because of the risk for immunologic deterioration and disease progression. The aggressive approach is supported by the observation in multiple studies that suppression of plasma HIV RNA by antiretroviral therapy is easier to achieve and maintain at higher CD4+ T cell counts and lower levels of plasma viral load (6,33--36). However, long-term clinical outcome data are not available to fully endorse this approach. Data are conflicting regarding sex-specific differences in viral load and CD4+ T cell counts. Certain studies (37--43), although not others (44--47), have concluded that after adjustment for CD4+ T cell count, levels of HIV RNA are lower in women than men. In those studies that have indicated a possible sex difference in HIV RNA levels, women have had RNA levels that ranged from 0.13 to 0.28 log10 lower than observed among men. In two studies of HIV seroconverters, HIV RNA copy numbers were substantially lower in women than men at seroconversion, but these differences decreased with time; and median viral load in women and men became similar within 5--6 years after seroconversion (38,39,43). Other data indicate that CD4+ T cell counts might be higher in women than men (48). However, importantly, rates of disease progression do not differ in a sex-dependent manner (41,43,49,50). Taken together, these data demonstrate that sex-based differences in viral load occur predominantly during a window of time when the CD4+ T cell count is relatively preserved, when treatment is recommended only in the setting of increased levels of plasma HIV RNA. Clinicians might consider lower plasma HIV RNA thresholds for initiating therapy in women with CD4+ T cell counts >350 cells/mm3, although insufficient data exist to determine an appropriate threshold. In patients with CD4+ T cell counts <350 cells/mm3, limited sex-based differences in viral load have been observed; therefore, no changes in treatment guidelines for women are recommended for this group. In summary, the decision to begin therapy for the asymptomatic patient with >200 CD4+ T cells/mm3 is complex and must be made in the setting of careful patient counseling and education. Factors that must be considered in this decision are 1) the willingness, ability, and readiness of the person to begin therapy; 2) the degree of existing immunodeficiency as determined by the CD4+ T cell count; 3) the risk for disease progression as determined by the CD4+ T cell count and level of plasma HIV RNA (1) (Figure; Tables 5,6); 4) the potential benefits and risks of initiating therapy for asymptomatic persons, including short- and long-term adverse drug effects (Table 4); and 5) the likelihood, after counseling and education, of adherence to the prescribed treatment regimen. Regarding adherence, no patient should automatically be excluded from consideration for antiretroviral therapy simply because he or she exhibits a behavior or other characteristic judged by the clinician to lend itself to nonadherence. Rather, the likelihood of patient adherence to a long-term, complex drug regimen should be discussed and determined by the patient and clinician before therapy is initiated. To achieve the level of adherence necessary for effective therapy, providers are encouraged to use strategies for assessing and assisting adherence: intensive patient education and support regarding the critical need for adherence should be provided; specific goals of therapy should be established and mutually agreed upon; and a long-term treatment plan should be developed with the patient. Intensive follow-up should occur to assess adherence to treatment and to continue patient counseling for the prevention of sexual and drug-injection--related transmission (see Adherence to Potent Antiretroviral Therapy). Considerations for Discontinuing TherapyAs recommendations evolve, patients who had begun active antiretroviral therapy at CD4+ T cell counts of >350/mm3 might consider discontinuing treatment. No clinical data exist addressing whether this should be done or if it can be accomplished safely. Potential benefits include reduction of toxicity and drug interactions, decreased risk for drug-selecting resistant variants, and improvement in quality of life. Risks include rebound in viral replication and renewed immunologic deterioration. If the patient and clinician agree to discontinue therapy, the patient should be closely monitored. Adherence to Potent Antiretroviral TherapyThe Panel recommends that certain persons living with HIV, including persons who are asymptomatic, should be treated with HAART for the rest of their lives. Adherence to the regimen is essential for successful treatment and has been reported to increase sustained virologic control, which is critical in reducing HIV-related morbidity and mortality. Conversely, suboptimal adherence has been reported to decrease virologic control and has been associated with increased morbidity and mortality (51,52). Suboptimal adherence also leads to drug resistance, limiting the effectiveness of therapy (53). The determinants, measurements, and interventions to improve adherence to HAART are insufficiently characterized and understood, and additional research regarding this topic is needed. Adherence to Therapy During HIV DiseaseAdherence is a key determinant in the degree and duration of virologic suppression. Among studies reporting on the association between suboptimal adherence and virologic failure, nonadherence among patients on HAART was the strongest predictor for failure to achieve viral suppression below the level of detection (52,53). Other studies have reported that 90%--95% of doses must be taken for optimal suppression, with lesser degrees of adherence being associated with virologic failure (51,54). No conclusive evidence exists that the degree of adherence required varies with different classes of agents or different medications in the HAART regimen. Suboptimal adherence is common. Surveys have determined that one third of patients missed doses within <3 days of the survey (55). Reasons for missed doses were predictable and included forgetting, being too busy, being out of town, being asleep, being depressed, having adverse side effects, and being too ill (56). One fifth of HIV-infected patients in one urban center never filled their prescriptions. Although homelessness can lead to suboptimal adherence, one program achieved a 70% adherence rate among homeless persons by using flexible clinic hours, accessible clinic staff, and incentives (57). Predictors of inadequate adherence to HIV medications include 1) lack of trust between the clinician and patient; 2) active drug and alcohol use; 3) active mental illness (e.g., depression); 4) lack of patient education and inability of patients to identify their medications (56); and 5) lack of reliable access to primary medical care or medication (58). Other sources of instability influencing adherence include domestic violence and discrimination (58). Medication side effects can also cause inadequate adherence as can fear of or experiencing metabolic and morphologic side effects of HAART (59). Predictors of optimal adherence to HIV medications, and hence, optimal viral suppression, include 1) availability of emotional and practical life supports; 2) a patient's ability to fit medications into his or her daily routine; 3) understanding that suboptimal adherence leads to resistance; 4) recognizing that taking all medication doses is critical; 5) feeling comfortable taking medications in front of others (60); and 6) keeping clinic appointments (34). Measurement of adherence is imperfect and lacks established standards. Patient self-reporting is an unreliable predictor of adherence; however, a patient's estimate of suboptimal adherence is a strong predictor and should be strongly considered (60,61). A clinician's estimate of the likelihood of a patient's adherence is also an unreliable predictor (62). Aids for measuring adherence (e.g., pill counts, pharmacy records, smart pill bottles with computer chips that record each opening [i.e., medication event monitoring systems or MEMS caps]) might be useful, although each aid requires comparison with patient self-reporting (61,63). Clinician and patient estimates of the degree of adherence have been reported to exceed measures that are based on MEMS caps. Because of its complexity and cost, MEMS caps technology might be used as an adjunct to adherence research, but it is not useful in clinical settings. Self-reporting should include a short-term assessment of each dose that was taken during the recent past (e.g., <3 days) and a general inquiry regarding adherence since the last visit, with explicit attention to the circumstances of missed doses and possible measures to prevent further missed doses. Having patients bring their medications and medication diaries to clinic visits might be helpful also. Approaching the PatientPatient-Related Strategies The first principle of patient-related strategies is to negotiate a treatment plan that the patient understands and to which he or she commits (Tables 7--10) (64,65). Before writing the first prescription, clinicians should assess the patient's readiness to take medication, which might take two or three office visits and patience. Patient education should include the goals of therapy, including a review of expected outcomes that are based on baseline viral load and CD4+ T cell counts (e.g., MACS data from the Guidelines [4]), the reason for adherence, and the plan for and mechanics of adherence. Patients must understand that the first HAART regimen has the best chance for long-term success (1). Clinicians and health teams should develop a plan for the specific regimen, including how medication timing relates to meals and daily routines. Centers have offered practice sessions and have used candy in place of pills to familiarize the patient with the rigors of HAART; however, no data exist to indicate if this exercise improves adherence. Daily or weekly pillboxes, timers with alarms, pagers, and other devices can be useful. Because medication side effects can affect treatment adherence, clinicians should inform patients in advance of possible side effects and when they are likely to occur. Treatment for side effects should be included with the first prescription, as well as instructions on appropriate response to side effects and when to contact the clinician. Low literacy is associated with suboptimal adherence, also. Clinicians should assess a patient's literacy level before relying on written information, and they should tailor the adherence intervention for each patient. Visual aids and audio or video information sources can be useful for patients with low literacy (66). Education of family and friends and their recruitment as participants in the adherence plan can be useful. Community interventions, including adherence support groups or the addition of adherence concerns to other support group agendas, can aid adherence. Community-based case managers and peer educators can assist with adherence education and strategies for each patient. Temporary postponement of HAART initiation has been proposed for patients with identified risks for suboptimal adherence (67,68). For example, a patient with active substance abuse or mental illness might benefit from psychiatric treatment or treatment for chemical dependency before initiating HAART. During the 1--2 months needed for treatment of these conditions, appropriate HIV therapy might be limited to OI prophylaxis, if indicated, and therapy for drug withdrawal, detoxification, or the underlying mental illness. In addition, readiness for HAART can be assessed and adherence education can be initiated during this period. Other sources of patient instability (e.g., homelessness) can be addressed during this time. Patients should be informed and in agreement with plans for future treatment and time-limited treatment deferral. Selected factors (e.g., sex, race, low socioeconomic status or education level, and past drug use) are not reliable predictors of suboptimal adherence. Conversely, higher socioeconomic status and education level and a lack of past drug abuse do not predict optimal adherence (69). No patient should automatically be excluded from antiretroviral therapy simply because he or she exhibits a behavior or characteristic judged by the clinician to indicate a likelihood of nonadherence. Clinician and Health Team-Related Strategies Trusting relationships among the patient, clinician, and health team are essential (Table 8). Clinicians should commit to communication between clinic visits, ongoing adherence monitoring, and timely response to adverse events or interim illness. Interim management during clinician vacations or other absences must be clarified with the patient. Optimal adherence requires full participation by the health-care team, with goal reinforcement by >2 team members. Supportive and nonjudgmental attitudes and behaviors will encourage patient honesty regarding adherence and problems. Improved adherence is associated with interventions that include pharmacist-based adherence clinics (69), street-level drop-in centers with medication storage and flexible hours for homeless persons (70), adolescent-specific training programs (71), and medication counseling and behavioral interventions (72) (Table 9). For all health-care team members, specific training regarding HAART and adherence should be offered and updated periodically. Monitoring can identify periods of inadequate adherence. Evidence indicates that adherence wanes as time progresses, even among patients whose adherence has been optimal, a phenomenon described as pill fatigue or treatment fatigue (67,73). Thus, monitoring adherence at every clinic encounter is essential. Reasonable responses to decreasing adherence include increasing the intensity of clinical follow-up, shortening the follow-up interval, and recruiting additional health team members, depending on the problem (68). Certain patients (e.g., chemically dependent patients, mentally retarded patients in the care of another person, children and adolescents, or patients in crisis) might require ongoing assistance from support team members from the outset. New diagnoses or symptoms can influence adherence. For example, depression might require referral, management, and consideration of the short- and long-term impact on adherence. Cessation of all medications at the same time might be more desirable than uncertain adherence during a 2-month exacerbation of chronic depression. Responses to adherence interventions among specific groups have not been well-studied. Evidence exists that programs designed specifically for adolescents, women and families, injection-drug users, and homeless persons increase the likelihood of medication adherence (69,71,74,75). The incorporation of adherence interventions into convenient primary care settings; training and deployment of peer educators, pharmacists, nurses, and other health-care personnel in adherence interventions; and monitoring of clinician and patient performance regarding adherence are beneficial (70,76,77). In the absence of data, a reasonable response is to address and monitor adherence during all HIV primary care encounters and incorporate adherence goals in all patient treatment plans and interventions. This might require the full use of a support team, including bilingual providers and peer educators for non-English--speaking populations, incorporation of adherence into support group agendas and community forums, and inclusion of adherence goals and interventions in the work of chemical-dependency counselors and programs. Regimen-Related Strategies Regimens should be simplified as much as possible by reducing the number of pills and therapy frequency and by minimizing drug interactions and side effects. For certain patients, problems with complex regimens are of lesser importance, but evidence supports simplified regimens with reduced pill numbers and dose frequencies (78,79). With the effective options for initial therapy noted in this report and the observed benefit of less frequent dosing, twice-daily dosing of HAART regimens is feasible for the majority of patients. Regimens should be chosen after review and discussion of specific food requirements and patient understanding and agreement to such restrictions. Regimens requiring an empty stomach multiple times daily might be difficult for patients with a wasting disorder, just as regimens requiring high fat intake might be difficult for patients with lactose intolerance or fat aversion. However, an increasing number of effective regimens do not have specific food requirements. Directly Observed TherapyDirectly observed therapy (DOT), in which a health-care provider observes the ingestion of medication, has been successful in tuberculosis management, specifically among patients whose adherence has been suboptimal. However, DOT is labor-intensive, expensive, intrusive, and programmatically complex to initiate and complete; and unlike tuberculosis, HIV requires lifelong therapy. Pilot programs have studied DOT among HIV patients with preliminary success. These programs have studied once-daily regimens among prison inmates, methadone program participants, and other patient cohorts with a record of repeated suboptimal adherence. Modified DOT programs have also been studied in which the morning dose is observed and evening and weekend doses were self-administered. The goal of these programs is to improve patient education and medication self-administration during a limited period (e.g., 3--6 months); however, the outcome of these programs, including long-term adherence after DOT completion, has not been determined (80--83). Therapy GoalsEradication of HIV infection cannot be achieved with available antiretroviral regimens, chiefly because the pool of latently infected CD4+ T cells is established during the earliest stages of acute HIV infection (84) and persists with a long half-life, even with prolonged suppression of plasma viremia to <50 copies/mL (85--88). The primary goals of antiretroviral therapy are maximal and durable suppression of viral load, restoration and preservation of immunologic function, improvement of quality of life, and reduction of HIV-related morbidity and mortality (Table 10). In fact, adoption of treatment strategies recommended in this report has resulted in substantial reductions in HIV-related morbidity and mortality (89--91). Plasma viremia is a strong prognostic indicator in HIV infection (3). Furthermore, reductions in plasma viremia achieved with antiretroviral therapy account for substantial clinical benefits (92). Therefore, suppression of plasma viremia as much as possible for as long as possible is a critical goal of antiretroviral therapy, but this goal must be balanced against the need to preserve effective treatment options. Switching antiretroviral regimens for any detectable level of plasma viremia can rapidly exhaust treatment options; reasonable parameters that can prompt a change in therapy are discussed in Criteria for Changing Therapy. HAART often leads to increases in the CD4+ T cell count of >100--200 cells/mm3, although patient responses are variable. CD4+ T cell responses are usually related to the degree of viral load suppression (93). Continued viral load suppression is more likely for those patients who achieve higher CD4+ T cell counts during therapy (94). A favorable CD4+ T cell response can occur with incomplete viral load suppression and might not indicate an unfavorable prognosis (95). Durability of the immunologic responses that occur with suboptimal suppression of viremia is unknown; therefore, although viral load is the strongest single predictor of long-term clinical outcome, clinicians should consider also sustained rises in CD4+ T cell counts and partial immune restoration. The urgency of changing therapy in the presence of low-level viremia is tempered by this observation. Expecting that continuing the existing therapy will lead to rapid accumulation of drug-resistant virus might not be reasonable for every patient. A reasonable strategy is maintenance of the regimen, but with redoubled efforts at optimizing adherence and increased monitoring. Partial reconstitution of immune function induced by HAART might allow elimination of unnecessary therapies (e.g., therapies used for prevention and maintenance against OIs). The appearance of naïve T cells (96,97), partial normalization of perturbed T cell receptor Vb repertoires (98), and evidence of residual thymic function in patients receiving HAART (99,100) demonstrate that partial immune reconstitution occurs in these patients. Further evidence of functional immune restoration is the return during HAART of in vitro responses to microbial antigens associated with opportunistic infections (101) and the lack of Pneumocystis carinii pneumonia among patients who discontinued primary Pneumocystis carinii pneumonia prophylaxis when their CD4+ T cell counts rose to >200 cells/mm3 during HAART (102--104). Guidelines include recommendations concerning discontinuation of prophylaxis and maintenance therapy for certain OIs when HAART-induced increases in CD4+ T cell counts occur (2). Tools for Achieving Therapy GoalsAlthough approximately 70%--90% of antiretroviral drug-naïve patients achieve maximal viral load suppression 6--12 months after therapy initiation, only 50% of patients in certain city clinics achieved similar results (33,34). Predictors of virologic success include low baseline viremia and high baseline CD4+ T cell count (33--35), rapid decline of viremia (6), decline of viremia to <50 HIV RNA copies/mL (6), adequate serum levels of antiretroviral drugs (6,105), and adherence to the drug regimen (34,51,106). Although optimal strategies for achieving antiretroviral therapy goals have not been fully delineated, efforts to improve patient adherence to therapy are critical (see Adherence to Potent Antiretroviral Therapy). Another tool for maximizing benefits of antiretroviral therapy is the rational sequencing of drugs and the preservation of future treatment options for as long as possible. Three alternative regimens include a protease inhibitor (PI) with two nucleoside reverse transcriptase inhibitors (NRTIs), a nonnucleoside reverse transcriptase inhibitor (NNRTI) with two NRTIs, or a 3-NRTI regimen (Table 11). The goal of a class-sparing regimen is to preserve or spare >1 classes of drugs for later use. Extending the overall long-term effectiveness of the available therapy options might be possible by sequencing drugs in this manner. Moreover, this strategy enables selectively delaying the risk for certain side effects associated with a single class of drugs. The efficacy of PI-containing HAART regimens has been reported to include durable viral load suppression, partial immunologic restoration, and decreased incidence of AIDS and death (24--26). Viral load suppression and CD4+ T cell responses that are similar to those observed with PI-containing regimens have been achieved with selected PI-sparing regimens (e.g., efavirenz plus two NRTIs [107] or abacavir plus two NRTIs [108]); however, whether such PI-sparing regimens will provide comparable efficacy with regard to clinical outcomes is unknown. The presence of drug-resistant HIV among treatment-experienced patients is a strong predictor of virologic failure and disease progression (109--111). Results of prospective studies indicate that the virologic response to a new antiretroviral regimen can be substantially improved when results of previous resistance testing are available to guide drug choices (10,11). Thus, resistance testing is a useful tool in selecting active drugs when changing antiretroviral regimens after virologic failure (see Drug-Resistance Testing). Initiating Therapy for the Asymptomatic HIV-Infected PatientWhen initiating antiretroviral therapy for the patient who is naïve to such therapy, clinicians should begin with a regimen that is expected to achieve sustained suppression of plasma HIV RNA, a sustained increase in CD4+ T cell count, and a favorable clinical outcome (i.e., delayed progression to AIDS and death). Clinicians should consider also the regimen's pill burden, dosing frequency, food requirements, convenience, toxicity, and drug-interaction profile compared with other regimens. Strongly recommended regimens include either indinavir, nelfinavir, ritonavir plus saquinavir; ritonavir plus indinavir; ritonavir plus lopinavir; or efavirenz in combination with one of the two NRTI combinations (Table 12). Clinical outcome data support using a PI in combination with NRTIs (24--26) (BI). Ritonavir as the single PI should be considered as an alternative agent because certain patients have difficulty tolerating standard doses of ritonavir (34) and because of the drug's multiple interactions. A similar rationale applies to saquinavir soft-gel capsule because certain patients have difficulty tolerating standard doses and because of the pill burden associated with its use; however, switching a patient off a ritonavir or saquinavir-based regimen is not necessary if they are tolerating the regimen and it is effective. Using ritonavir to increase plasma concentrations of other PIs has evolved from an investigational concept to widespread practice. Standard doses of PIs result in trough drug levels (i.e., the lowest drug levels in the patient's system) that are only slightly higher than the effective antiviral concentration, which could allow viral replication. In contrast, protease boosting or enhancement by administering ritonavir increases the trough levels of other PIs higher than the IC50 or IC95, which minimizes opportunities for viral replication and potentially allows drug activity against even moderately resistant strains of virus. Additionally, these dual-PI combinations can lead to more convenient regimens regarding pill burden, scheduling, and elimination of food restrictions. They also might prevent efavirenz- or nevirapine-induced drug interactions. Ritonavir increases plasma concentrations of other PIs by >2 mechanisms, including inhibition of gastrointestinal cytochrome P450 (CYP450) during absorption, and metabolic inhibition of hepatic CYP450. The 20-fold increase in saquinavir plasma concentrations with ritonavir coadministration is probably caused by CYP450 inhibition at both sites and leads to an increase in the saquinavir peak plasma concentration (Cmax) (112). For lopinavir, the addition of ritonavir increases the Cmax and half-life, which subsequently results in a higher trough concentration. The result is a lopinavir blood concentration curve that is 100-fold higher, compared with lopinavir alone (113). For other PIs, metabolism in the gastrointestinal tract is less critical, and the enhancement is primarily the result of CYP450 inhibition in the liver. The addition of ritonavir to amprenavir, nelfinavir, or indinavir results in substantial increases in half-life and trough levels, with a more moderate or minimal increase in Cmax (114,115). The dose of ritonavir that is used for PI boosting is also critical for certain PIs but not others. With saquinavir and amprenavir, increases in the ritonavir dose to >100 mg two times/day do not significantly increase the PI levels (114,116). However, increasing ritonavir doses to >100 mg two times/day provides additional enhancement for indinavir and nelfinavir (115,117). Although pharmacokinetic data support using ritonavir-plus-PI combinations, limited data are available regarding combinations other than ritonavir plus saquinavir (118) or ritonavir plus lopinavir (119). In addition, the long-term risks and toxicities of dual-PI combinations remain unknown. Disappointing results with antiretroviral regimens prescribed after virologic failure with a previous regimen indicate that the first regimen affords the best opportunity for long-term control of viral replication. Because the genetic barrier to resistance is greatest with PIs, experienced clinicians consider a PI plus two NRTIs to be the preferred initial regimen. However, efavirenz plus two NRTIs is as effective as one PI plus two NRTIs in suppressing plasma viremia and increasing CD4+ T cell counts (107), and certain experienced clinicians prefer this as the initial regimen because it might spare the toxicities of PIs for a substantial time (BII). Although no direct comparative trials have been reported that would allow a ranking of relative efficacy of NNRTIs, the ability of efavirenz in combination with two NRTIs to suppress viral replication and increase CD4+ T cell counts to a similar degree as one PI with two NRTIs supports a preference for efavirenz over other presently available NNRTIs. Abacavir plus two NRTIs, a 3-NRTI regimen, has been used successfully as well (108) (CII). However, such a regimen might have short-lived efficacy when the baseline viral load is >100,000 copies/mL. Using two NRTIs does not achieve the goal of suppressing viremia to below detectable levels as consistently as does a regimen in the strongly recommended or alternative categories and should be used only if more potent treatment is impossible (DI). Use of antiretroviral agents as monotherapy is contraindicated (DI), except when no other options exist or during pregnancy to reduce perinatal transmission. When initiating antiretroviral therapy, all drugs should be started simultaneously at full dose with the following exceptions: dose escalation regimens are recommended for ritonavir, nevirapine, and for certain patients, ritonavir plus saquinavir. Hydroxyurea has been used investigationally in combination with antiretroviral agents for treatment of HIV infection; however, its utility in this setting has not been established. Clinicians considering use of hydroxyurea in a treatment regimen for HIV should be aware of the limited and conflicting nature of data in support of its efficacy and the importance of monitoring patients closely for potentially serious toxicity.** Detailed information is included in this report comparing NRTIs, NNRTIs, PIs, drug interactions between PIs and other agents, toxicities, and FDA-required warning labels (Tables 13--20). Drug interactions between PIs and other agents can be extensive and often require dose modification or substitution of different drugs (Tables 17--19). Toxicity assessment is an ongoing process; assessment >2 times during the first month of therapy and every 3 months thereafter is a reasonable management approach. Initiating Therapy for the Patient with Advanced HIV DiseaseAll patients with diagnosed advanced HIV disease, which is defined as any condition meeting the 1993 CDC definition of AIDS (23), should be treated with antiretroviral agents, regardless of plasma viral levels (AI). All patients with symptomatic HIV infection without AIDS (i.e., the presence of thrush or unexplained fever) should be treated also. When the patient is acutely ill with an OI or other complication of HIV infection, the clinician should consider clinical problems (e.g., drug toxicity, ability to adhere to treatment regimens, drug interactions, or laboratory abnormalities) when determining the timing of antiretroviral therapy initiation. When therapy is initiated, a maximally suppressive regimen should be used (Table 12). Advanced stage patients being maintained on an antiretroviral regimen should not discontinue therapy during an acute OI or malignancy, unless drug toxicity, intolerance, or drug interactions are of concern. When patients who have progressed to AIDS are treated with complicated combinations of drugs, potential multidrug interactions must be appreciated by the clinician and patient. Thus, when choosing antiretroviral agents, the clinician should consider potential drug interactions and overlapping drug toxicities (Tables 13--20). For example, using rifampin to treat active tuberculosis is problematic for a patient receiving a PI that adversely affects the metabolism of rifampin but might be needed to effectively suppress viral replication. Conversely, rifampin lowers the blood level of PIs, which can result in suboptimal antiretroviral therapy. Although rifampin is contraindicated or not recommended for use with all PIs, clinicians can consider using rifabutin at a reduced dose (Table 18); this topic is discussed in detail elsewhere (120). Other factors complicating advanced disease are wasting and anorexia disorders, which can prevent patients from adhering to the dietary requirements for efficient absorption of certain PIs. Bone marrow suppression associated with zidovudine and the neuropathic effects of zalcitabine, stavudine, and didanosine can combine with the direct effects of HIV to render the drugs intolerable. Hepatotoxicity associated with certain PIs and NNRTIs can limit the use of these drugs (e.g., for patients with underlying liver dysfunction). The absorption and half-life of certain drugs can be altered by antiretroviral agents, including PIs and NNRTIs whose metabolism involves CYP450 enzymatic pathway. PIs inhibit the CYP450 pathway, whereas NNRTIs have variable effects. Nevirapine is an inducer; delavirdine is an inhibitor; and efavirenz is a mixed inducer and inhibitor. CYP450 inhibitors can increase blood levels of drugs metabolized by this pathway. Adding a CYP450 inhibitor can improve the pharmacokinetic profile of selected agents (e.g., adding ritonavir therapy to saquinavir) as well as contribute an antiviral effect; however, these interactions can also result in life-threatening drug toxicity (Tables 11--19). Thus, clinicians should discuss with their patients any new drugs, including over-the-counter and alternative medications, that the patient might consider taking. Relative risks versus benefits of specific combinations of agents should be considered. Initiation of potent antiretroviral therapy is associated with degrees of immune function recovery. Patients with advanced HIV disease and subclinical OIs (e.g., Mycobacterium avium-intracellulare or cytomegalovirus) can experience a new immunologic response to the pathogen, and thus, new symptoms can occur in association with the heightened immunologic or inflammatory response. This response should not be interpreted as an antiretroviral therapy failure, and these new OIs should be treated appropriately while maintaining the patient on the antiretroviral regimen. Viral load measurement is helpful in clarifying the patient's condition. HAART-Associated Adverse Clinical EventsLactic Acidosis and Hepatic SteatosisChronic compensated hyperlactatemia can occur during treatment with NRTIs (121,122). Although cases of severe decompensated lactic acidosis with hepatomegaly and steatosis are rare (estimated incidence of 1.3 cases/1,000 person-years of NRTI exposure), this syndrome is associated with a high mortality rate (123--126). Severe lactic acidosis with or without pancreatitis, including three fatal cases, were reported during the later stages of pregnancy or among postpartum women whose antiretroviral therapy during pregnancy included stavudine and didanosine in combination with other antiretroviral agents (125,127,128). Other risk factors for experiencing this toxicity include obesity, being female, and prolonged use of NRTIs, although cases have been reported with risk factors being unknown (125). The mitochondrial basis of NRTI-induced lactic acidosis and hepatic steatosis is one possible mechanism of cellular injury because NRTIs also inhibit deoxyribonucleic acid (DNA) polymerase gamma, which is the enzyme responsible for mitochondrial DNA synthesis. The ensuing mitochondrial dysfunction might also result in multiple other adverse events (e.g., pancreatitis, peripheral neuropathy, myopathy, and cardiomyopathy) (129). Certain features of lipodystrophy syndrome have been hypothesized as being tissue-specific mitochondrial toxicities caused by NRTI treatment (130--132). The initial clinical signs and symptoms of patients with lactic acidosis syndrome are variable and can include nonspecific gastrointestinal symptoms without substantial elevation of hepatic enzymes (133). Clinical prodromes can include otherwise unexplained onset and persistence of abdominal distention, nausea, abdominal pain, vomiting, diarrhea, anorexia, dyspnea, generalized weakness, ascending neuromuscular weakness, myalgias, paresthesias, weight loss, and hepatomegaly (134). In addition to hyperlactatemia, laboratory evaluation might reveal an increased anion gap (Na -- [Cl + CO2] > 16), elevated aminotransferases, creatine phosphokinase, lactic dehydrogenase, lipase, and amylase (124,133,135). Echotomography and computed tomography (CT) scans might indicate an enlarged fatty liver, and histologic examination of the liver might reveal microvesicular steatosis (133). Because substantial technical problems are associated with lactate testing, routine monitoring of lactate level is not usually recommended. Clinicians must first rely on other laboratory abnormalities plus symptoms when lactic acidosis is suspected. Measurement of lactate requires a standardized mode of sample handling, including prechilled fluoride-oxalate tubes, which should be transported immediately on ice to the laboratory and processed within 4 hours after collection; blood should be collected without using a tourniquet, without fist-clenching, and if possible, without stasis (136,137). When interpreting serum lactate, levels of 2--5 mmol/dL are considered elevated and need to be correlated with symptoms. Levels >5 mmol/dL are abnormal, and levels >10 mmol/dL indicate serious and possibly life-threatening situations. Certain persons knowledgeable in HIV treatment also recommend monitoring of serum bicarbonate and electrolytes for the early identification of an increased anion gap every 3 months. For certain patients, the adverse event resolves after discontinuation of NRTIs (133,138), and they tolerate administration of a revised NRTI-containing regimen (133,139); however, insufficient data exist to recommend this strategy versus treatment with an NRTI-sparing regimen. If NRTI treatment is continued, for certain patients, progressive mitochondrial toxicity can produce severe lactic acidosis manifested clinically by tachypnea and dyspnea. Respiratory failure can follow, requiring mechanical ventilation. In addition to discontinuation of antiretroviral treatment and intensive therapeutic strategies that include bicarbonate infusions and hemodialysis (140) (AI), clinicians can administer thiamine (141) and riboflavin (127) on the basis of the pathophysiologic hypothesis that sustained cellular dysfunctions of the mitochondrial respiratory chain cause this fulminant clinical syndrome. However, efficacy of these latter interventions requires clinical validation. Antiretroviral treatment should be suspended if clinical and laboratory manifestations of the lactic acidosis syndrome occur (BIII). HepatotoxicityHepatotoxicity, which is defined as a 3--5 times increase in serum transaminases (e.g., aspartate aminotransferase, alanine aminotransferase, or gamma-glutamyltransferase) with or without clinical hepatitis, has been reported among patients receiving HAART. All marketed NNRTIs and PIs have been associated with serum transaminase elevation. The majority of patients are asymptomatic, and certain cases resolve spontaneously without therapy interruption or modification (142). Hepatic steatosis in the presence of lactic acidosis is a rare but serious adverse effect associated with the nucleoside analogs (see more detailed discussion in Lactic Acidosis and Hepatic Steatosis). Among the NNRTIs, nevirapine has the greatest potential for causing clinical hepatitis. An incidence of 12.5% of hepatotoxicity among patients initiating nevirapine has been reported, with clinical hepatitis diagnosed for 1.1% of these patients (143). In an African randomized trial where stavudine was the backbone NRTI, and either nevirapine or efavirenz was added to emtricitabine or lamivudine, 9.4% of the nevirapine-treated patients experienced grade 4 liver enzyme elevation as compared with none of the efavirenz-treated patients. Two of these patients died of liver failure. The incidence among female patients was twice that observed among male patients (12% versus 6%; p = 0.05) (144). Nevirapine-associated hepatitis might also be present as part of a hypersensitivity syndrome, with a constellation of other symptoms (e.g., skin rash, fever, and eosinophilia). Approximately two thirds of the cases of nevirapine-associated clinical hepatitis occur within the first 12 weeks. Fulminant and even fatal cases of hepatic necrosis have been reported. Patients might experience nonspecific gastrointestinal and flu-like symptoms with or without liver enzyme abnormalities. The syndrome can progress rapidly to hepatomegaly, jaundice, and hepatic failure within days (145). A two-week lead-in dosing with 200 mg once daily before dose escalation to twice daily might reduce the incidence of hepatotoxicity. Because of the potential severity of clinical hepatitis, certain clinicians advise close monitoring of liver enzymes and clinical symptoms after nevirapine initiation (e.g., every 2 weeks for the first month; then monthly for first 12 weeks, and every 1--3 months thereafter). Patients who experience severe clinical hepatotoxicity while receiving nevirapine should not receive nevirapine therapy in the future. Unlike the early onset hepatotoxicity observed with nevirapine, PI-associated liver enzyme abnormalities can occur any time during the treatment course. In a retrospective review, severe hepatotoxicity (defined as a >5 times increase over baseline aspartate aminotransferase or alanine aminotransferase) was observed more often among patients receiving ritonavir- or ritonavir/saquinavir-containing regimens than those receiving indinavir, nelfinavir, or saquinavir (146). Coinfection with hepatitis C virus is reported to be a major risk factor for development of hepatotoxicity after PI initiation (147,148). HAART-induced immune reconstitution rather than direct liver toxic effects of the PIs have been indicated as the cause of liver decompensation among hepatitis C or hepatitis B coinfected patients. Other potential risk factors for hepatotoxicity include hepatitis B infection (142,147,149), alcohol abuse (148), baseline elevated liver enzymes (150), stavudine use (149), and concomitant use of other hepatotoxic agents. HyperglycemiaHyperglycemia, new-onset diabetes mellitus, diabetic ketoacidosis, and exacerbation of preexisting diabetes mellitus have been reported among patients receiving HAART (151--153). These metabolic derangements are strongly associated with PI use (154), although they can occur independent of PI use (155). The incidence of new onset hyperglycemia was reported as 5% in a 5-year historical cohort analysis of a population of 221 HIV-infected patients. PIs were independently associated with hyperglycemia, and the incidence did not vary substantially by PIs (156). Viral load suppression and increase in body weight did not reduce the magnitude of the association with PIs. The pathogenesis of these abnormalities has not been fully elucidated; however, hyperglycemia might result from peripheral and hepatic insulin resistance, relative insulin deficiency, an impaired ability of the liver to extract insulin, and a longer exposure to antiretroviral medications (157,158). Hyperglycemia with or without diabetes has been reported among 3%--17% of patients in multiple retrospective studies. In these reports, symptoms of hyperglycemia were reported at a median of approximately 60 days (range: 2--390 days) after initiation of PI therapy. Hyperglycemia resolved for certain patients who discontinued PI therapy; however, the reversibility of these events is unknown because of limited data. Certain patients continued PI therapy and initiated treatment with oral hypoglycemic agents or insulin. Clinicians are advised to monitor closely their HIV-infected patients with preexisting diabetes when PIs are prescribed and to be aware of the risk for drug-related new-onset diabetes among patients without a history of diabetes (BIII). Patients should be advised regarding the warning signs of hyperglycemia (i.e., polydipsia, polyphagia, and polyuria) and the need to maintain a recommended body weight when these medications are prescribed. Certain clinicians recommend routine fasting blood glucose measurements at 3--4-month intervals during the first year of PI treatment for patients with no previous history of diabetes (CIII). Routine use of glucose tolerance tests to detect this complication is not recommended (DIII). Because pregnancy is an independent risk factor for impaired glucose tolerance, closer monitoring of blood glucose levels should be done for pregnant women receiving PI-containing regimens. No data are available to aid in the decision to continue or discontinue drug therapy among patients with new-onset or worsening diabetes; however, the majority of experienced clinicians recommend continuation of HAART in the absence of severe diabetes (BIII). Studies have attempted to examine the potential of reversing insulin resistance after switching from PI-containing HAART regimens to NNRTI-based regimens, but results have been inconclusive. Fat MaldistributionHIV infection and antiretroviral therapy have been associated with unique fat distribution abnormalities. Generalized fat wasting is common in advanced HIV disease, and localized fat accumulations have been reported with NRTI monotherapy (159). However, the recognition and observation of fat maldistribution syndromes have increased in the era of combination antiretroviral therapy characterized by fat wasting (lipoatrophy) or fat accumulation (hyperadiposity). Fat maldistribution is often referred to as lipodystrophy, and in combination with metabolic abnormalities including insulin resistance and hyperlipidemia is referred to as lipodystrophy syndrome. The absence of a commonly used case definition for the different forms of lipoatrophy or fat accumulation, often collectively called lipodystrophy, has lead to different prevalence estimates (range: 25%--75%) (160--163). Although the lack of defining criteria has also impeded investigation into the pathogenic mechanisms of these abnormalities, the spectrum of morphologic abnormalities might indicate multifactorial causation related to specific antiretroviral exposure and underlying host factors. Lipodystrophy might be associated with serum dyslipidemias, glucose intolerance, or lactic acidosis (163--165). Fat accumulation might be seen in the abdomen, the dorsocervical fat pad, and, among both men and women, the breasts. Prevalence increases with duration of antiretroviral therapy (166). Although available evidence indicates that an increased risk for fat accumulation exists with PIs, whether specific drugs are more strongly associated with this toxicity is unclear. The face and extremities are most commonly affected by fat atrophy, and variability exists in severity. Prevalence of this toxicity has been reported to increase with long-term NRTI exposure (167). Although stavudine has been frequently reported in cases of lipoatrophy, this might be a marker of longer term treatment exposure (168--171). No clearly effective therapy for fat accumulation or lipoatrophy is known. In the majority of persons, discontinuation of antiretroviral medications or class switching has not resulted in substantial benefit; however, among a limited number of persons, improvement in physical appearance has been reported (172). Preliminary results from limited studies indicate a reduction in accumulated fat and fat redeposition with the use of certain agents (personal communication, M. Schambelan and P.A. Volberding, 2001) (173). However, data are inconclusive and recommendations cannot be made. HyperlipidemiaHIV infection and antiretroviral therapy are associated with complex metabolic alterations, including dyslipidemia. Cachexia, reduced total cholesterol, and elevated triglycerides were reported before the availability of potent antiretroviral therapy (174,175). HAART is associated with elevation of total serum cholesterol and low-density lipoprotein and in additional increases in fasting triglycerides (162,176). The magnitude of changes varies substantially and does not occur among all patients. Dyslipidemias primarily occur with PIs; however, a range from an increased association with ritonavir to limited or no association with a newer investigational compound indicates that hyperlipidemia might be a drug-specific rather than a class-specific toxicity (177). Frequently, antiretroviral-associated dyslipidemias are sufficiently severe enough to consider therapeutic intervention. Although data remain inconclusive, lipid elevations might be associated with accelerated atherosclerosis and cardiovascular complications among HIV-infected persons. Indications for monitoring and intervention in HIV therapy-associated dyslipidemias are the same as among uninfected populations (178). No evidence-based guidelines exist for lipid management specific to HIV infection and antiretroviral therapy. However, close monitoring of lipid levels among patients with additional risks for atherosclerotic disease might be indicated (179). Low-fat diets, regular exercise, control of blood pressure and smoking cessation are critical elements of care. Hypercholesterolemia might respond to b-hdroxy-b-methylglutaryl-CoA reductase inhibitors (statins). However, recognizing the interactions of certain statins with PIs that can result in increased statin levels is critical (Table 17). Usually, agents that are less affected by the inhibitory effect of PIs via the cytochrome P450 system are preferred (e.g. pravastatin). Atorvastatin, which is at least partially metabolized by this pathway, can also be used with PIs. However, atorvastatin should be used with caution and at reduced doses because higher concentrations of atorvastatin are expected (180). Monotherapy with fibrates is less effective, but they can be added to statin therapy; additional monitoring is needed because of the increased risk of rhabdomyolysis and hepatotoxicity. Isolated triglyceride elevations respond best to low-fat diets, fibrates, or statins (180,181). Lipid elevations might require modifications in antiretroviral regimens if they are severe or unresponsive to other management strategies. Numerous trials, variably well-controlled, have demonstrated modest reductions in lipid elevations when an NNRTI replaces a PI or when an abacavir-containing triple NRTI regimen replaces a PI-containing regimen (182--184). Improvement in lipid levels tends to be more substantial with nevirapine than with efavirenz in studies regarding switching therapies. Increased Bleeding Episodes Among Patients with HemophiliaIncreased spontaneous bleeding episodes among patients with hemophilia A and B have been observed with PI use (185). Reported episodes have involved joints and soft tissues; however, serious bleeding episodes, including intracranial and gastrointestinal bleeding, have been reported. Bleeding episodes occurred a median of 22 days after initiation of PI therapy. Certain patients received additional coagulation factor while continuing PI therapy. Osteonecrosis, Osteopenia, and OsteoporosisAvascular necrosis and decreased bone density are now recognized as emerging metabolic complications of HIV infection that might be linked to HAART regimens. Both of these bone abnormalities have been reported among adults and children with HIV infection who are now surviving longer with their disease in part because of HAART (186--188). Avascular necrosis involving the hips was first described among HIV-infected adults and more recently among HIV-infected children (known as Legg-Calvé-Perthes disease). Diagnoses of osteonecrosis are usually made by CT scan or magnetic resonance imaging (MRI), when these studies are performed in response to patient's complaints of pain in an affected hip or spine. However, asymptomatic disease with MRI findings can occur among 5% of HIV patients (189). Avascular necrosis is not associated with a specific antiretroviral regimen among HIV-infected adults, but it has been linked to corticosteroids use among certain patients (189,190). Factors associated with osteonecrosis include alcohol abuse, hemoglobinopathies, corticosteroid treatment, hyperlipidaemia, and hypercoagulability states. Occurrence of hyperlipidaemia indicates an indirect link between antiretroviral therapy and the occurrence of osteonecrosis among HIV-infected patients; however, prospective clinical studies are required to establish this association. No accepted medical therapy exists for avascular necrosis, and surgery might be necessary for disabling symptoms. Decreases in bone mineral density (BMD), both moderate (osteopenia) and severe (osteoporosis), are a reflection of the competing effects of bone reabsorption by osteoclast and bone deposition by osteoblast and are measured by bone densitometry. Before HAART, marginal decreases in BMD among HIV-infected persons were reported (191). This evidence for decreased bone formation and turnover has been demonstrated with more potent antiretroviral therapy, including PIs (192). Studies of bone demineralization among a limited number of patients receiving HAART have reported that <50% of patients receiving a PI-based regimen experienced osteopenia, compared with 20% of patients who are untreated or receiving a non-PI--containing regimen (193). Other studies have reported that patients with lipodystrophy with extensive prior PI therapy had associated findings of osteopenia (28%) or osteoporosis (9%), respectively (194). Preliminary observations of increased serum and urinary markers of bone turnover among patients on protease-containing HAART who have osteopenia support the possible link of bone abnormalities to other metabolic abnormalities observed among HIV-infected patients (195,196). Presently, no recommendation can be made for routine measurement of bone density among asymptomatic patients by dual energy X-ray absorptiometry (DEXA) or by such newer measurements as quantitative ultrasound (QUS). Specific prophylaxis or treatment recommendations to prevent more substantial osteoporosis have not been developed for HIV-infected patients with osteopenia. On the basis of experience in the treatment of primary osteoporosis, recommending adequate intake of calcium and vitamin D and appropriate weight-bearing exercise is reasonable. When fractures occur or osteoporosis is documented, more specific and aggressive therapies with bisphosphonates, raloxifene, or calcitonin might be indicated (197). Hormone replacement therapy including estrogen can be considered in the setting of substantially decreased bone density among postmenopausal women on HAART. Skin RashSkin rash occurs most commonly with the NNRTI class of drugs. The majority of cases are mild to moderate, occurring within the first weeks of therapy. Certain experienced clinicians recommend managing the skin rash with antihistamine for symptomatic relief without drug discontinuation, although continuing treatment during such rashes has been questioned (198). More serious cutaneous manifestations (e.g., Stevens-Johnson syndrome [SJS] and toxic epidermal necrosis [TEN]) should result in the prompt and permanent discontinuation of NNRTI or other offending agents. The majority of reactions resulting in skin rash are confined to cutaneous reactions. However, a severe or even life-threatening syndrome of drug rash with eosinophilia and systemic symptoms (DRESS) has also been described (199,200). Systemic symptoms can include fever, hematological abnormalities, and multiple organ involvement. Among NNRTIs, skin rash occurs more frequently and in greater severity with nevirapine. Using a 2-week lead-in dose escalation schedule when initiating nevirapine therapy might reduce the incidence of rash. In a case-control multinational study, SJS and TEN were reported among 18 HIV-infected patients. Fifteen of the 18 patients were receiving nevirapine. The median time from initiation of nevirapine to onset of cutaneous eruption was 11 days, with two thirds of the cases occurring during the initial dosing period (198). Female patients might have as much as a sevenfold higher risk for developing grade 3 or 4 skin rashes than male patients (201,202). The use of systemic corticosteroid or antihistamine therapy at the time of the initiation of nevirapine to prevent development of skin rash has not proven effective (202,203). In fact, a higher incidence of skin rash has been reported among the steroid- or antihistamine-treated patients. At present, prophylactic use of corticosteroids should be discouraged. Skin rash appears to be a class-adverse reaction of the NNRTIs. The incidence of cross-hypersensitivity reactions between these agents is unknown. In a limited number of reports, patients with prior history of nevirapine-associated skin rash had been able to tolerate efavirenz without increased rates of cutaneous reactions (204,205). The majority of experienced clinicians do not recommend using another NNRTI among those patients who experienced SJS or TEN with one NNRTI. Initiating NNRTI for a patient with a history of mild to moderate skin rash with another NNRTI should be done with caution and close follow-up. Among the NRTIs, skin rash occurs most frequently with abacavir. Skin rash might be one of the symptoms of abacavir-associated systemic hypersensitivity reaction; in that case, therapy should be discontinued without future attempts to resume abacavir therapy. Among all PIs, skin rash occurs most frequently with amprenavir, with incidence of <27% in clinical trials. Although amprenavir is a sulfonamide, the potential of cross-reactivity between amprenavir and other sulfa drugs is unknown. As a result, amprenavir should be used with caution in patients with history of sulfa allergies. Interruption of Antiretroviral TherapyAntiretroviral therapy might need to be discontinued temporarily or permanently for multiple reasons. If a need exists to discontinue any antiretroviral medication, clinicians and patients should be aware of the theoretical advantage of stopping all antiretroviral agents simultaneously, rather than continuing one or two agents, to minimize the emergence of resistant viral strains. If a decision is made to interrupt therapy, the patient should be monitored closely, including clinical and laboratory evaluations. Chemoprophylaxis against OIs should be initiated as needed on the basis of CD4+ T cell count. An interest exists in what is sometimes referred to as structured or supervised treatment interruptions (STI). The concepts underlying STI vary, depending on patient populations, and encompass >3 major strategies: 1) STI as part of salvage therapy; 2) STI for autoimmunization and improved immune control of HIV; and 3) STI for the sole purpose of allowing less total time on antiretroviral therapy. Because of limited available data, none of these approaches can be recommended. Salvage STI is intended for patients whose virus has developed substantial antiretroviral drug resistance and who have persistent plasma viremia and relatively low CD4+ T cell counts despite receiving therapy. The theoretical goal of STI in this patient population is to allow for the reemergence of HIV that is susceptible to antiretroviral therapy. Although HIV that was sensitive to antiretroviral agents was detected in the plasma of persons after weeks or months of interrupted treatment, the emergence of drug-sensitive HIV was associated with a substantial decline in CD4+ T cells and a substantial increase in plasma viremia, indicating improved replicative fitness and pathogenicity of wild type virus (206). In addition, drug-resistant HIV persisted in CD4+ T cells. The observed decrease in CD4+ T cells is of concern in this patient population, and STI cannot be recommended for these patients. Autoimmunization STI and STI for the reduction of total time receiving antiretroviral drugs are intended for persons who have maintained suppression of plasma viremia below the limit of detection for prolonged periods of time and who have relatively high CD4+ T cell counts. The theoretical goal of autoimmunization STI is to allow multiple short bursts of viral replication to augment HIV-specific immune responses. This strategy is being studied among persons who began HAART during either the very early or chronic stages of HIV infection (207--209). STI for the purpose of less time on therapy uses predetermined periods of long- or short-cycle intermittent antiretroviral therapy. The numbers of patients and duration of follow-up are insufficient for adequate evaluation of these approaches. Risks include a decline in CD4+ T cell counts, an increase in transmission, and the development of drug resistance. Because of insufficient data regarding these situations, STI cannot be recommended for use in general clinical practice. Further research is necessary in each of these areas. Changing a Failing RegimenAs with the initiation of antiretroviral therapy, deciding to change regimens should be approached after considering multiple, complex factors, including
A regimen can fail for multiple reasons, including among other reasons, initial viral resistance to >1 agents, altered absorption or metabolism of the drug, multidrug pharmacokinetics that adversely affect therapeutic drug levels, and inadequate patient adherence to a regimen. Careful assessment of a patient's adherence before changing antiretroviral therapy is critical; the patient's other health-care providers (e.g., the case manager or social worker) can assist with this evaluation. Clinicians should be aware of the prevalence of mental health disorders and psychoactive substance use disorders among HIV-infected persons because suboptimal mental health treatment services can jeopardize the ability of these persons to adhere to medical treatment. Optimal identification of and intervention for these mental health disorders can enhance adherence to HIV therapy. Clinicians should distinguish between drug failure versus drug toxicity before changing a patient's therapy. In cases of drug toxicity, >1 alternative drugs of the same potency and from the same class of agents as the suspected agent should be substituted. In cases of drug failure where >2 drugs have been used, a detailed history of current and past antiretroviral medications, as well as other HIV-related medications, should be obtained. Testing for antiretroviral drug resistance can also be helpful in maximizing the number of active drugs in a regimen (see Drug-Resistance Testing). Viral resistance to antiretroviral drugs can be a key reason for treatment failure. Genetically distinct viral variants emerge in each HIV-infected person after initial infection. Viruses with single-drug--resistant mutations exist even before therapy but are selected for replication by antiviral regimens that are only partially suppressive. The more potent a regimen is in durably suppressing HIV replication, the less probable the emergence of resistant variants. Thus, a therapy's goal should be to reduce plasma HIV RNA to below detectable limits (i.e., <50 copies/mL), thereby providing the strongest possible genetic barrier to drug resistance. Three groups of patients should be considered for a change in therapy: 1) persons who are receiving incompletely suppressive antiretroviral therapy (e.g., single- or double-nucleoside therapy) with detectable or undetectable plasma viral load; 2) persons who have been on potent combination therapy and whose viremia was initially suppressed to undetectable levels but has again become detectable; and 3) persons who have been on potent combination therapy and whose viremia was never suppressed to below detectable limits. Criteria for Changing TherapyThe goal of antiretroviral therapy, to improve the length and quality of patients' lives, is best accomplished by maximal suppression of viral replication to below detectable levels (i.e., <50 copies/mL) sufficiently early to preserve immune function. However, to achieve this goal for certain patients, therapy regimens must be modified. Plasma HIV RNA level is the key parameter for evaluating therapy response, and increases in levels of viremia that are substantial, confirmed, and not attributable to intercurrent infection or vaccination, indicate failure of the drug regimen regardless of changes in the CD4+ T cell counts. Clinical complications and sequential changes in CD4+ T cell count can complement the viral load test in evaluating a treatment response. Specific criteria that should prompt consideration for changing therapy include the following:
A final consideration is the recognition of the limited choice of available agents and the knowledge that a decision to change regimens might reduce future treatment options for that patient. This consideration can influence the clinician to be more conservative when deciding to change therapy. Consideration of alternative options should include potency of the substituted regimen and probability of tolerance of, or adherence to, the alternative regimen. Clinical trials have demonstrated that partial suppression of virus is superior to no suppression of virus. Conversely, clinicians and patients might prefer to suspend treatment to preserve future options or because a sustained antiviral effect cannot be achieved. Referral to or consultation with an experienced HIV clinician is appropriate when considering a change in therapy. When possible, patients requiring a change in antiretroviral regimen, but who are without treatment options through using approved drugs, should be referred for inclusion in a clinical trial. Therapeutic Options When Changing Antiretroviral TherapyRecommendations for changes in treatment differ according to the indication for the change. If the desired virologic objectives have been achieved for patients who have intolerance or toxicity, a substitution for the offending drug should be made, preferably by using an agent in the same class with a different toxicity or tolerance profile. If virologic objectives have been achieved, but the patient is receiving a regimen not in the preferred category (e.g., two NRTIs or monotherapy), treatment can be continued with careful monitoring of viral load, or drugs can be added to the current regimen to comply with strongly recommended treatment regimens. As previously discussed, the majority of experienced clinicians believe that treatment with regimens not in the strongly recommended or alternative categories is associated with eventual failure, and they recommend the latter tactic. Limited clinical data exist to support specific strategies for changing therapy among patients who have failed the strongly recommended regimens; however, theoretical considerations should guide decisions. Because of the rapid mutability of HIV, viral strains with resistance to >1 agents can emerge during therapy, chiefly when viral replication has not been maximally suppressed. Of concern is the possibility of broad cross-resistance among drugs within a class. Evidence indicates that viral strains that become resistant to one PI or NNRTI often have reduced susceptibility to the majority or all other PIs or NNRTIs. A change in regimen because of treatment failure should be guided by results of resistance testing. This report includes a summary of the guidelines to follow when changing a patient's antiretroviral therapy (Table 21). Dose modifications might be necessary to account for drug interactions when using combinations of PIs or a PI and NNRTI (Table 19). For certain patients, options are limited because of previous antiretroviral use, toxicity, or intolerance. For the clinically stable patient with detectable viremia for whom an optimal change in therapy is not possible, delaying therapy changes in anticipation of the availability of newer and more potent agents might be prudent. Decisions to change therapy and design a new regimen should be made with assistance from a clinician well-experienced in treating HIV-infected patients through consultation or referral. Acute HIV InfectionAn estimated 40%--90% of patients acutely infected with HIV will experience certain symptoms of acute retroviral syndrome (Table 22) and should be considered for early therapy (210--213). However, acute HIV infection is often not recognized by primary care clinicians because of the similarity of the symptom complex with those of influenza or other illnesses. Additionally, acute primary infection can occur asymptomatically. Health-care providers should consider a diagnosis of HIV infection for patients who experience a compatible clinical syndrome (Table 22) and should obtain appropriate laboratory testing. Evidence includes detectable HIV RNA in plasma by using sensitive PCR or bDNA assays combined with a negative or indeterminate HIV antibody test. Although measurement of plasma HIV RNA is the preferable diagnostic method, a test for p24 antigen might be useful when RNA testing is not readily available. However, a negative p24 antigen test does not eliminate acute infection, and a low titer (<10,000 copies/mL), false-positive test can exist with HIV RNA levels. When suspicion for acute infection is high (e.g., in a patient with a report of recent risk behavior in association with the symptoms and signs listed in Table 22), a test for HIV RNA should be performed (BII). Patients with diagnosed HIV infection by HIV RNA testing should have confirmatory testing performed (Table 2). Information regarding treatment of acute HIV infection from clinical trials is limited. Preliminary data indicate that treatment of primary HIV infection with combination therapy has a beneficial effect on laboratory markers of disease progression (19,214--216). However, the potential disadvantages of initiating therapy include additional exposure to antiretroviral therapy without a known clinical benefit, which could result in substantial toxicities, development of antiretroviral drug resistance, and adverse effect on quality of life. Ongoing clinical trials are addressing the question of long-term benefits of potent treatment regimens. Theoretically, early intervention can
The potential risks of therapy for acute HIV infection include
These considerations are similar to those for initiating therapy for the asymptomatic patient (see Considerations for Initiating Therapy for the Patient with Asymptomatic HIV-Infection). The health-care provider and the patient should be aware that therapy of primary HIV infection is based on theoretical considerations, and the potential benefits should be weighed against the potential risks. Certain authorities endorse treatment of acute HIV infection on the basis of the theoretical rationale and limited but supportive clinical trial data. Apart from patients with acute primary HIV infection, experienced clinicians also recommend consideration of therapy for patients among whom seroconversion has occurred within the previous 6 months (CIII). Although the initial burst of viremia among infected adults usually resolves in 2 months, treatment during the 2--6-month period after infection is based on the probability that virus replication in lymphoid tissue is still not maximally contained by the immune system during this time (217). Decisions regarding therapy for patients who test antibody-positive and who believe the infection is recent, but for whom the time of infection cannot be documented, should be made by using the algorithm discussed in Considerations for Patients with Established HIV Infection (CIII). Except for postexposure prophylaxis with antiretroviral agents (218), no patient should be treated for HIV infection until the infection has been documented. All patients being examined without a formal medical record of a positive HIV test (e.g., those who have a positive result from a home test kit) should undergo enzyme-linked immunosorbent assay and an established confirmatory test (e.g., Western Blot) to document HIV infection (AI). Treatment Regimen for Primary HIV InfectionAfter the clinician and patient have made the decision to use antiretroviral therapy for primary HIV infection, treatment should be implemented in an attempt to suppress plasma HIV RNA levels to below detectable levels (AIII). Data are insufficient to draw firm conclusions regarding specific drug recommendations; potential combinations of agents available are similar to those used in established infection (Table 12). These aggressive regimens can be associated with disadvantages, including drug toxicity, pill burden, cost, and the possibility of drug resistance that could limit future options. The latter is probable if virus replication is not adequately suppressed or if the patient has been infected with a viral strain that is already resistant to one or more agents. The patient should be counseled regarding potential limitations, and decisions should be made only after weighing the risks and sequelae of therapy against the theoretical treatment benefits. Because 1) the goal of therapy is suppression of viral replication to below the level of detection; 2) the benefits of therapy are based on theoretical considerations; and 3) long-term clinical outcome benefit has not been documented, any regimen that is not expected to maximally suppress viral replication is not appropriate for treating the acutely HIV-infected person (EIII). Additional clinical studies are needed to delineate the role of antiretroviral therapy during the primary infection period. Patient Follow-UpTesting for plasma HIV RNA levels and CD4+ T cell count and toxicity monitoring should be performed as described in Testing for Plasma HIV RNA Levels and CD4+ T Cell Count To Guide Decisions Regarding Therapy (i.e., on initiation of therapy, after 4 weeks, and every 3--4 months thereafter) (AII). However, certain experienced clinicians believe that testing for plasma HIV RNA levels at 4 weeks is not helpful in evaluating the therapy's effect regarding acute infection, because viral loads might be decreasing from peak viremia levels, even in the absence of therapy. Duration of Therapy for Primary HIV InfectionAfter therapy is initiated, experienced clinicians recommend continuing treatment with antiretroviral agents indefinitely because viremia has been documented to reappear or increase after therapy discontinuation (CII). Optimal duration and therapy composition are unknown, but ongoing clinical trials should provide relevant data regarding these concerns. Difficulties inherent in determining the optimal duration and therapy composition initiated for acute infection should be considered when first counseling the patient regarding therapy. Considerations for Antiretroviral Therapy Among HIV-Infected AdolescentsHIV-infected adolescents who were infected through sex or injection-drug use during adolescence follow a clinical course that is more similar to HIV disease among adults than children. In contrast, adolescents who were infected perinatally or through blood products as young children have a unique clinical course that differs from other adolescents and long-term surviving adults. The majority of HIV-infected adolescents were infected through sex during the adolescent period and are in an early stage of infection. Puberty is a time of somatic growth and hormone-mediated changes, with females acquiring additional body fat and males additional muscle mass. Theoretically, these physiologic changes can affect drug pharmacology, including drugs with a narrow therapeutic index that are used in combination with protein-bound medicines or hepatic enzyme inducers or inhibitors. However, no clinically substantial impact of puberty has been reported with NRTI use. Clinical experience with PIs and NNRTIs has been limited. Thus, medication dosages used to treat HIV and OIs among adolescents should be based on Tanner staging of puberty and not specific age. Adolescents in early puberty (Tanner stages I and II) should be administered dosages on the basis of pediatric guidelines, whereas those in late puberty (Tanner stage V) should be administered dosages on the basis of adult guidelines. Youth who are in the midst of their growth spurt (Tanner stage III females and Tanner stage IV males) should be monitored closely for medication efficacy and toxicity when choosing adult or pediatric dosing guidelines. Considerations for Antiretroviral Therapy Among HIV-Infected Pregnant WomenAntiretroviral treatment recommendations for HIV-infected pregnant women are based on the belief that therapies of known benefit to women should not be withheld during pregnancy, unless the risk for adverse effects outweighs expected benefits for the woman. Combination antiretroviral therapy is the recommended standard treatment for HIV-infected nonpregnant women. Additionally, a three-part regimen of zidovudine, administered orally starting at 14 weeks gestation and continued throughout pregnancy, intravenously during labor and to the newborn for the first 6 weeks of life, reduced the risk for perinatal transmission by 66% in a randomized, double-blind clinical trial (i.e., the Pediatric AIDS Clinical Trials Group [PACTG] protocol 076) (20) and is recommended for all pregnant women (219). Pregnancy should not preclude the use of optimal therapeutic regimens. However, recommendations regarding choices of antiretroviral drugs for treatment of infected women are subject to unique considerations including 1) potential changes in dosing requirement resulting from physiologic changes associated with pregnancy, 2) potential effects of antiretroviral drugs on a pregnant woman, 3) effect on the risk for perinatal HIV transmission, and 4) the potential short- and long-term effects of the antiretroviral drug on the fetus and newborn, all of which might not be known for certain antiretroviral drugs (219). The decision to use any antiretroviral drug during pregnancy should be made by the woman after discussion with her clinician regarding the benefits versus risks to her and her fetus. Long-term follow-up is recommended for all infants born to women who have received antiretroviral drugs during pregnancy. Women who are in the first trimester of pregnancy and who are not receiving antiretroviral therapy might wish to consider delaying therapy initiation until after 10--12 weeks gestation. This period of organogenesis is when the embryo is most susceptible to potential teratogenic drug effects, and the risks regarding antiretroviral therapy to the fetus during that period are unknown. However, this decision should be discussed between the clinician and patient and should include an assessment of the woman's health status and the benefits versus risks of delaying therapy initiation for these weeks. If clinical, virologic, or immunologic parameters are such that therapy would be recommended for nonpregnant women, the majority of Panel members recommend initiating therapy regardless of gestational age. Nausea and vomiting during early pregnancy, affecting the woman's ability to take and absorb oral medications, can be a factor in the decision regarding treatment during the first trimester. Standard combination antiretroviral therapy is recommended as initial therapy for HIV-infected pregnant women whose clinical, immunologic, or virologic status would indicate treatment if not pregnant. When antiretroviral therapy initiation would be considered optional on the basis of current guidelines for treatment of nonpregnant women, but HIV-1 RNA levels are >1,000 copies/mL, infected pregnant women should be counseled regarding the benefits of standard combination therapy and offered therapy, including the three-part zidovudine chemoprophylaxis regimen (Table 23). Although such women are at low risk for clinical disease progression if combination therapy is delayed, antiretroviral therapy that successfully reduces HIV-1 RNA levels to <1,000 copies/mL substantially lowers the risk for perinatal transmission (220--222) and limits the need to consider elective cesarean delivery as an intervention to reduce transmission risk (219). Use of antiretroviral prophylaxis has been demonstrated to provide benefit in preventing perinatal transmission, even for infected pregnant women with HIV-1 RNA levels <1,000 copies/mL. In a meta-analysis of factors associated with perinatal transmission among women who had infected infants despite having HIV-1 RNA <1,000 copies/mL at or near delivery, transmission was only 1.0% among women receiving zidovudine prophylaxis compared with 9.8% among those receiving no antiretroviral treatment (220). The time-limited use of zidovudine alone during pregnancy for chemoprophylaxis of perinatal transmission is controversial. Potential benefits of standard combination antiretroviral regimens for treatment of HIV infection should be discussed with and offered to all pregnant HIV-infected women regardless of viral load and is recommended for all pregnant women with HIV-1 RNA levels >1,000 copies/mL. However, a woman might wish to restrict exposure of her fetus to antiretroviral drugs during pregnancy but still wish to reduce the risk for transmitting HIV to her infant. Additionally, for women with HIV-1 RNA levels <1,000 copies/mL, time-limited use of zidovudine during the second and third trimesters of pregnancy is less likely to induce resistance caused by the limited viral replication existing in the patient and the time-limited exposure to the antiretroviral drug. For example, zidovudine resistance was unusual among healthy women who participated in PACTG 076 (21). Use of zidovudine chemoprophylaxis alone during pregnancy might be an appropriate option for these women. When combination therapy is administrated principally to reduce perinatal transmission and would have been considered optional for treatment if the woman were not pregnant, consideration can be given to discontinuing therapy postnatally, with the decision to reinstitute treatment on the basis of standard criteria for nonpregnant women. If drugs are discontinued postnatally, all drugs should be stopped simultaneously. Discussion regarding the decision to continue or stop combination therapy postpartum should occur before initiation of therapy during pregnancy. Women already receiving antiretroviral therapy might recognize their pregnancy early enough in gestation that concern for potential teratogenicity can lead them to consider temporarily stopping antiretroviral therapy until after the first trimester. Insufficient data exist to support or refute teratogenic risk regarding antiretroviral drug use among humans when administered during the first 10--12 weeks of gestation. However, treatment with efavirenz should be avoided during the first trimester because substantial teratogenic effects among rhesus macaques occurred at drug exposures similar to those representing human exposure. Hydroxyurea is a potent teratogen among animal species and should be avoided also during the first trimester. Temporary discontinuation of antiretroviral therapy could result in a rebound in viral levels that theoretically could be associated with increased risk for early in utero HIV transmission or could potentiate disease progression in the woman (223). Although the effects of all antiretroviral drugs on the developing fetus during the first trimester are uncertain, experienced clinicians recommend continuation of a maximally suppressive regimen, even during the first trimester. If antiretroviral therapy is discontinued during the first trimester for any reason, all agents should be stopped simultaneously to avoid drug resistance. After the drugs are reinstituted, they should be introduced simultaneously for the same reason. Limited data are available on the pharmacokinetics and safety of antiretroviral agents during pregnancy for drugs other than zidovudine.†† In the absence of data, drug choices should be personalized on the basis of discussion with the patient and available data from preclinical and clinical testing of each drug. FDA's pregnancy classification for all currently approved antiretroviral agents and selected other information regarding the use of antiretroviral drugs is available in this report (Table 24).The predictive value of in vitro and animal screening tests for adverse effects among humans is unknown. Certain drugs commonly used to treat HIV infection or its consequences can result in positive readings on >1 screening tests. For example, acyclovir is positive on certain in vitro assays for chromosomal breakage and carcinogenicity and is associated with fetal abnormalities among rats; however, data regarding human experience from the Acyclovir in Pregnancy Registry indicate no increased risk for birth defects among human infants with in utero exposure (224). When combination antiretroviral therapy is administered during pregnancy, zidovudine should be included as a component of antenatal therapy whenever possible. Circumstances might arise where this option is not feasible (e.g., occurrence of substantial zidovudine-related toxicity). Additionally, women receiving an antiretroviral regimen that does not contain zidovudine but who have HIV-1 RNA levels that are consistently low or undetectable have a low risk for perinatal transmission, and addition of zidovudine to the current regimen could compromise regimen adherence. Regardless of the antepartum antiretroviral regimen, intravenous intrapartum zidovudine and the standard 6-week course of zidovudine for the infant is recommended. If the woman has not received zidovudine as a component of her antenatal therapeutic antiretroviral regimen, intravenous zidovudine should still be administered to the pregnant woman during the intrapartum period, when feasible. Additionally, for women receiving combination antiretroviral treatment, the maternal antenatal antiretroviral treatment regimen should be continued on schedule as much as possible during labor to provide maximal virologic effect and to minimize the chance of drug resistance. Zidovudine and stavudine should not be administered together because of potential pharmacologic antagonism; therefore, options for women receiving oral stavudine as part of their antenatal therapy include continuing oral stavudine during labor without intravenous zidovudine or withholding oral stavudine during intravenous administration during labor. Toxicity related to mitochondrial dysfunction has been reported among HIV-infected patients receiving long-term treatment with nucleoside analogues and can be of concern for pregnant women. Symptomatic lactic acidosis and hepatic steatosis can have a female preponderance (125). Additionally, these syndromes have similarities to the rare but life-threatening syndromes of acute fatty liver of pregnancy and hemolysis, elevated liver enzymes and low platelets (HELLP syndrome) that occur during the third trimester of pregnancy. Certain data indicate that a disorder of mitochondrial fatty acid oxidation in the mother or her fetus during late pregnancy can affect the etiology of acute fatty liver of pregnancy and HELLP syndrome (225,226) and possibly contribute to susceptibility to antiretroviral-associated mitochondrial toxicity. Whether pregnancy augments the incidence of the lactic acidosis/hepatic steatosis syndrome reported among nonpregnant women receiving nucleoside analogue treatment is unclear. Bristol-Myers Squibb has reported three maternal deaths caused by lactic acidosis, two with and one without accompanying pancreatitis, among women who were either pregnant or postpartum and whose antepartum therapy during pregnancy included stavudine and didanosine in combination with other antiretroviral agents (either a PI or nevirapine) (128). All cases were among women who were receiving treatment with these agents at the time of conception and continued for the duration of pregnancy; all of the women were seen late in gestation with symptomatic disease that progressed to death in the immediate postpartum period. Two women were also associated with fetal demise. Nonfatal cases of lactic acidosis among pregnant women have also been reported. Because pregnancy itself can mimic certain early symptoms of lactic acidosis/hepatic steatosis syndrome or be associated with other disorders of liver metabolism, clinicians who care for HIV-infected pregnant women receiving nucleoside analogue drugs need to be alert for this syndrome. Pregnant women receiving nucleoside analogue drugs should have hepatic enzymes and electrolytes assessed more frequently during the last trimester of pregnancy, and any new symptoms should be evaluated thoroughly. Additionally, because of reports of maternal mortality secondary to lactic acidosis with prolonged use of the combination of stavudine and didanosine by HIV-infected pregnant women, clinicians should prescribe this antiretroviral combination during pregnancy with caution and only when other nucleoside analogue drug combinations have failed or caused unacceptable toxicity or side effects (128). The antenatal zidovudine dosing regimen used in the perinatal transmission prophylaxis trial PACTG 076 was zidovudine 100 mg administered five times/day and was selected on the basis of standard zidovudine dosage for adults at the time the study was designed in 1989 (Table 23). However, data indicate that administration of zidovudine three times/day will maintain intracellular zidovudine triphosphate at levels comparable with those observed with more frequent dosing (227,228). Comparable clinical response also has been observed in clinical trials among persons receiving zidovudine two times/day (229--231). Thus, the standard zidovudine dosing regimen for adults is 200 mg three times/day or 300 mg two times/day. A less-frequent dosing regimen would be expected to enhance maternal adherence to the zidovudine perinatal prophylaxis regimen and, therefore, is an acceptable alternative antenatal dosing regimen for zidovudine prophylaxis. In a short-course antenatal/intrapartum zidovudine perinatal transmission prophylaxis trial in Thailand, administration of zidovudine 300 mg two times/day for 4 weeks antenatally and 300 mg every 3 hours orally during labor was reported to reduce perinatal transmission by approximately 50%, compared with a placebo (232). The lower efficacy of the short-course two-part zidovudine prophylaxis regimen studied in Thailand compared with the three-part zidovudine prophylaxis regimen used in PACTG 076 and recommended for use in the United States, could result from 1) the shorter antenatal duration of zidovudine, 2) oral rather than intravenous administration during labor, 3) lack of treatment for the infant, or 4) a combination of these factors. In the United States, identification of HIV-infected pregnant women before or as early as possible during the course of pregnancy and use of the full three-part PACTG 076 zidovudine regimen is recommended for prevention of perinatal HIV transmission. Monitoring and use of HIV-1 RNA for therapeutic decision-making during pregnancy should be performed as recommended for nonpregnant women. Data from untreated and zidovudine-treated infected pregnant women indicate that HIV-1 RNA levels correlate with risk for transmission (20,221,222). However, although risk for perinatal transmission among women with HIV-1 RNA below the level of assay quantitation is low, transmission from mother to infant has been reported among women with all levels of maternal HIV-1 RNA. Additionally, antiretroviral prophylaxis is effective in reducing transmission even among women with low HIV RNA levels (20,220). Although the mechanism by which antiretroviral prophylaxis reduces transmission is probably multifactorial, reduction in maternal antenatal viral load is a key component of prophylaxis. However, pre- and postexposure prophylaxis of the infant is provided by passage of antiretroviral drugs across the placenta, resulting in inhibitory drug levels in the fetus during and immediately after the birth process (233). The extent of transplacental passage varies among antiretroviral drugs (Table 24). Additionally, although a correlation exists between plasma and genital tract viral load, discordance has also been reported (234--236). Further, differential evolution of viral sequence diversity occurs between the peripheral blood and genital tract (236,237). Studies are needed to define the relationship between viral load suppression by antiretroviral therapy in plasma and levels of HIV in the genital tract and the relationship between these compartment-specific effects and the risk for perinatal HIV transmission. The full zidovudine chemoprophylaxis regimen, including intravenous zidovudine during delivery and zidovudine administration to the infant for the first 6 weeks of life, in combination with other antiretrovirals or alone, should be discussed with and offered to all infected pregnant women regardless of their HIV-1 RNA level. Clinicians who are treating HIV-infected pregnant women are strongly encouraged to report cases of prenatal exposure to antiretroviral drugs (either administered alone or in combinations) to the Antiretroviral Pregnancy Registry. The registry collects observational, nonexperimental data regarding antiretroviral exposure during pregnancy for the purpose of assessing potential teratogenicity. Registry data will be used to supplement animal toxicology studies and assist clinicians in weighing the potential risks and benefits of treatment for each patient. The registry is a collaborative project with an advisory committee of obstetric and pediatric practitioners, staff from CDC and NIH, and staff from pharmaceutical manufacturers. The registry allows the anonymity of patients, and birth outcome follow-up is obtained by registry staff from the reporting clinician. Referrals should be directed to
Prevention Counseling for the HIV-Infected PatientOngoing prevention counseling is an essential component of management for HIV-infected persons (238). Each patient encounter provides an opportunity to reinforce HIV prevention messages. Therefore, each encounter should include assessment and documentation of 1) the patient's knowledge and understanding of HIV transmission and 2) the patient's HIV transmission behaviors since the last encounter with a member of the health-care team. This should be followed by a discussion of strategies to prevent transmission that might be useful to the patient. The physician, nurse, or other health-care team member should routinely provide this counseling. Partner notification is a key component of HIV detection and prevention and should be pursued by the provider or by referral services. Although the core elements of HIV prevention messages are unchanged since the introduction of HAART, key observations regarding the biology of HIV transmission, the impact of HAART on transmission, and personal risk behaviors have been noted. For example, sustained low plasma viremia that results from successful HIV therapy substantially reduces the likelihood of HIV transmission. In one study, for each log reduction in plasma viral load, the likelihood of transmission between discordant couples was reduced 2.5-fold (239). Similarly, mother-to-child HIV transmission was observed to decline in a linear fashion with each log reduction in maternal delivery viral load (221,222,238,239). Although this relationship is usually linear, key exceptions should be noted. For example, mother-to-child transmission has been reported even among women with very low or undetectable viral loads (220,240,241). Similarly, the relationship between viral load in the plasma and the levels in the genital fluid of women and the seminal fluid of men is complex. Studies have demonstrated a rough correlation between plasma HIV levels and genital HIV levels, but key exceptions have been observed (240). Viral evolution can occur in the genital compartment that is distinct from the viral evolution in the plasma, and transmissions have been documented in the presence of an undetectable plasma viral load (20,220,241). Thus, although durably effective HAART substantially reduces the likelihood of HIV transmission, the degree of protection is incomplete. Certain biologic factors other than plasma viral load have also been demonstrated to influence sexual transmission of HIV, including ulcerative and nonulcerative sexually transmitted infections (242), vaginitis (including bacterial vaginosis and Candida albicans vaginal infections) (243); genital irritation associated with frequent use of nonoxynol-9 (N-9)--containing products (244); menstruation; lack of circumcision in men (245--247); oral contraceptive use (248); estrogen deficiency (248); progesterone excess (243); and deficiencies of vitamin A (249) and selenium (247). Behavioral changes among HIV-infected persons have been observed during the HAART era that impact prevention. Unfortunately, evidence exists that awareness of the potential benefits of HAART is leading certain persons to relapse into high-risk activities. For example, reports from urban communities of men who have sex with men (MSM) in the United States indicate rising HIV seroprevalence rates, as well as rising rates of unsafe sexual practices, corroborated by the rising rates of other sexually transmitted infections. Recently, an association between knowledge of the benefits of HAART among MSM and relapse to high-risk activity was observed (250,251). Women might have unprotected sex because they wish to become pregnant. For women of childbearing potential, desire for pregnancy should be assessed at each encounter; women wishing to pursue pregnancy should be referred for preconception counseling to reduce risks for perinatal transmission and transmission to uninfected sexual partners. Among women of childbearing age who wish to avoid pregnancy, condoms should be encouraged in addition to other forms of contraception for preventing transmission of HIV and other sexually transmitted infections (dual method use) or used as a single method for pregnancy prevention as well (dual protection). In a randomized placebo-controlled clinical trial of N-9 conducted among commercial sex workers with high rates of sexual activity, N-9 did not protect against HIV infection, resulted in increased vaginal lesions, and possibly caused increased transmission (243). Although these adverse effects might not occur with less frequent use, given current evidence, spermicides containing N-9 should not be recommended as an effective means of HIV prevention. Optimal adherence with antiretroviral regimens has been directly associated with a lower risk for morbidity and mortality and indirectly with a reduction in risk for HIV transmission because of its association with lower viral loads (252). Suboptimal adherence to HIV medication recently has been demonstrated to be a predictor of suboptimal adherence to HIV prevention strategies (253). More intensive adherence and prevention counseling might be appropriate for persons who demonstrate repeated deficiencies in either area. Despite the strong association between a reduced risk for HIV transmission and sustained low viral load, the message of HIV prevention for patients should remain simple: After becoming infected, a person can transmit the virus at any time, and no substitute exists for latex or polyurethane male or female condoms, other safer sexual behaviors (e.g., partner reduction or abstinence), and cessation of any sharing of drug paraphernalia. Prevention counseling for patients known to have HIV infection remains a critical component of HIV primary care, including easy access to condoms and other means of prevention. Clinicians might wish to directly address with their patients the risks associated with using viral load outcomes as a factor in considering high-risk behavior. HIV-infected persons who use injection drugs should be advised to enroll in drug rehabilitation programs. If this advice is not followed or if these services are unavailable, the patient should receive counseling regarding risks associated with sharing needles and paraphernalia. Finally, the most successful and effective prevention messages are those tailored to each patient. These messages are culturally appropriate, practical, and relevant to the person's knowledge, beliefs, and behaviors (238). The message, the manner of delivery, and the cultural context vary substantially, depending on the patient (for additional information regarding these strategies, as well as recommendations on prevention, see HIV Prevention at http://hivinsite.ucsf.edu/InSite.jsp?page=kb-07). ConclusionThe Panel has attempted to use the advances in knowledge regarding the pathogenesis of HIV in the infected person to translate scientific principles and data obtained from clinical experience into guidelines that can be used by clinicians and patients to make therapeutic decisions. These guidelines are offered for ongoing discussion between the patient and clinician after having defined specific therapeutic goals with an acknowledgment of uncertainties. Patients should be entered into a continuum of medical care and services, including social, psychosocial, and nutritional services, with the availability of professional referral and consultation. To achieve the maximal flexibility in tailoring therapy to each patient during his or her infection, drug formularies must allow for all FDA-approved NRTIs, NNRTIs, and PIs as treatment options. The Panel urges industry and the public and private sectors to conduct further studies to allow refinement of these guidelines. Specifically, studies are needed to optimize recommendations for primary therapy; to define secondary therapy; and to delineate the reasons for treatment failure. The Panel remains committed to revising these guidelines as new data become available. References
* See inside back cover for list of panel members. † In this report, an adolescent is defined as a person in late puberty or stage V of the Tanner growth chart (i.e., sexually mature). § The panel's reports and updates are available from the HIV/AIDS Treatment Information Service (ATIS) (800-448-0440; TTY, 888-480-3739; or Fax, 301-519-6616) and on the ATIS Internet site at http://www.hivatis.org. They are also available from the National Prevention Information Network (NPIN) Internet site at http://www.cdcnpin.org. ¶ Additional information is available at http://hiv-web.lanl.gov. ** Additional information is available at http://www.hivatis.org. †† Additional information is available at http://www.hivatis.org. Table 1  Return to top. Figure 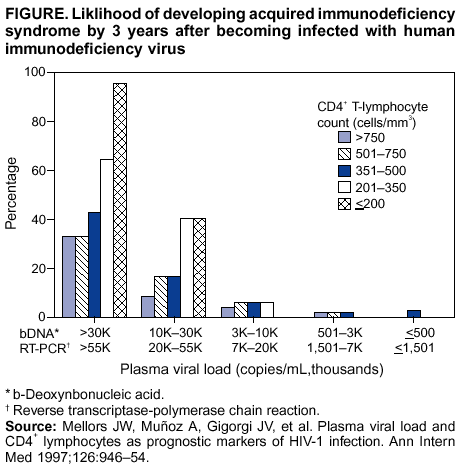 Return to top. Table 2  Return to top. Table 3 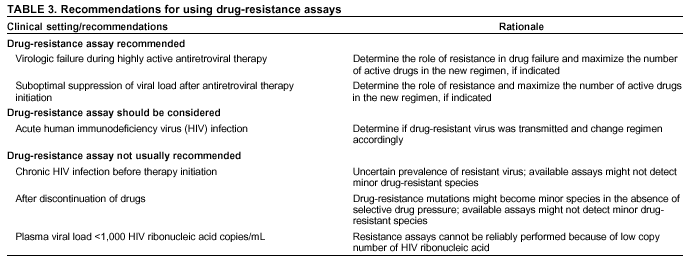 Return to top. Table 4  Return to top. Table 5 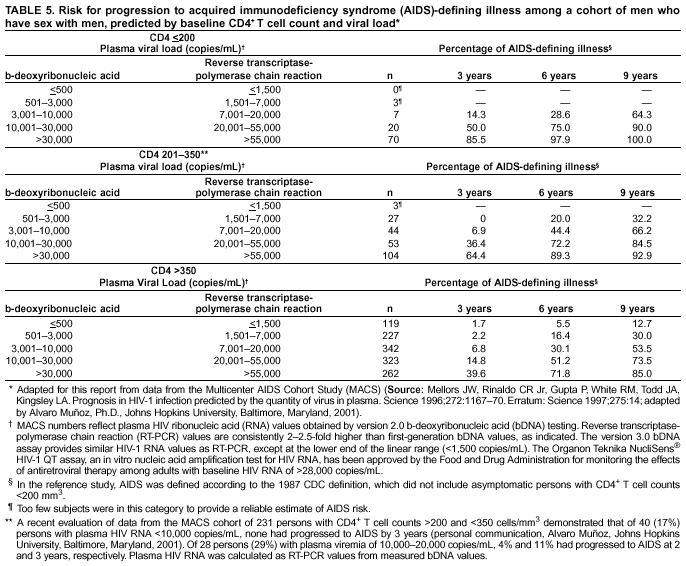 Return to top. Table 6  Return to top. Table 7  Return to top. Table 8  Return to top. Table 9  Return to top. Table 10  Return to top. Table 11 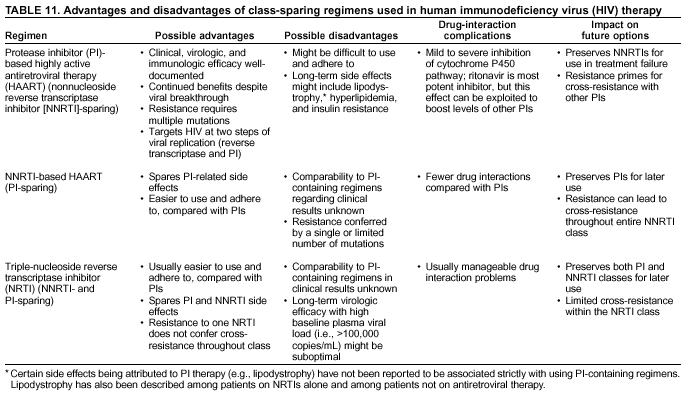 Return to top. Table 12  Return to top. Table 13 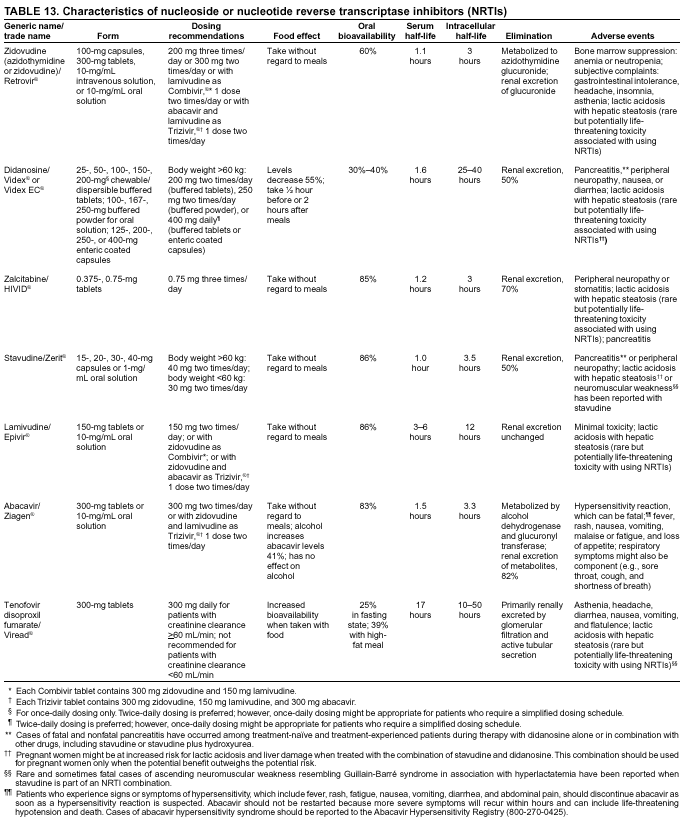 Return to top. Table 14 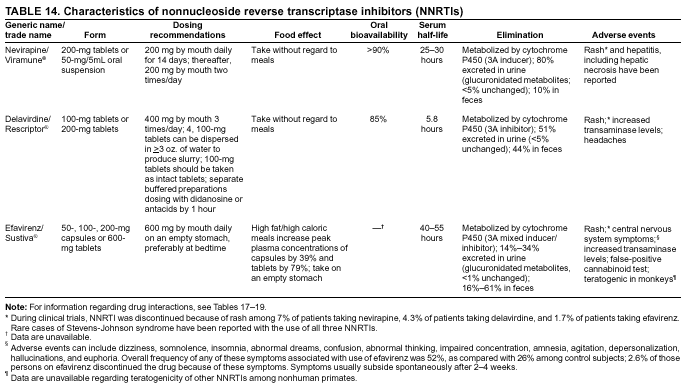 Return to top. Table 15 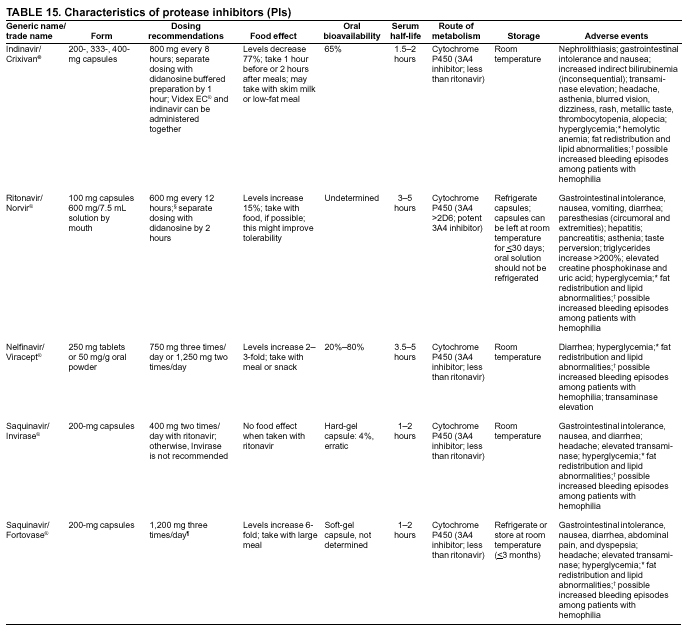 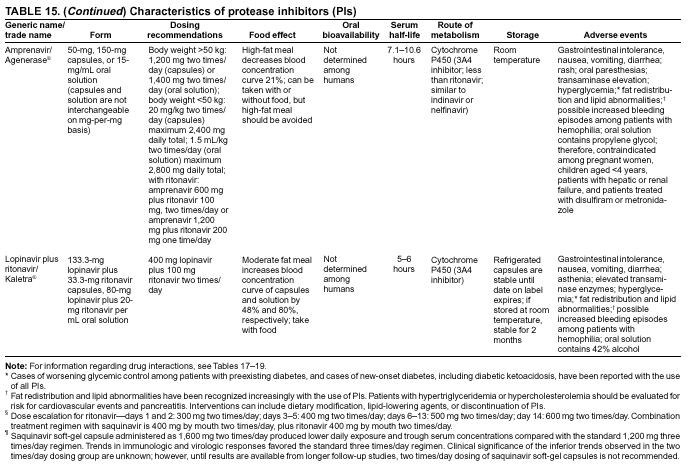 Return to top. Table 16 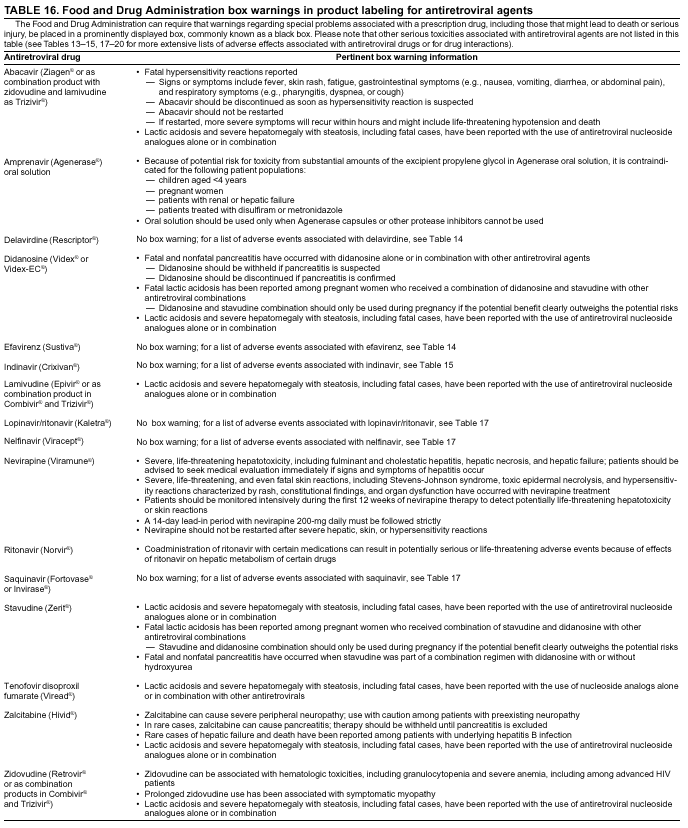 Return to top. Table 17 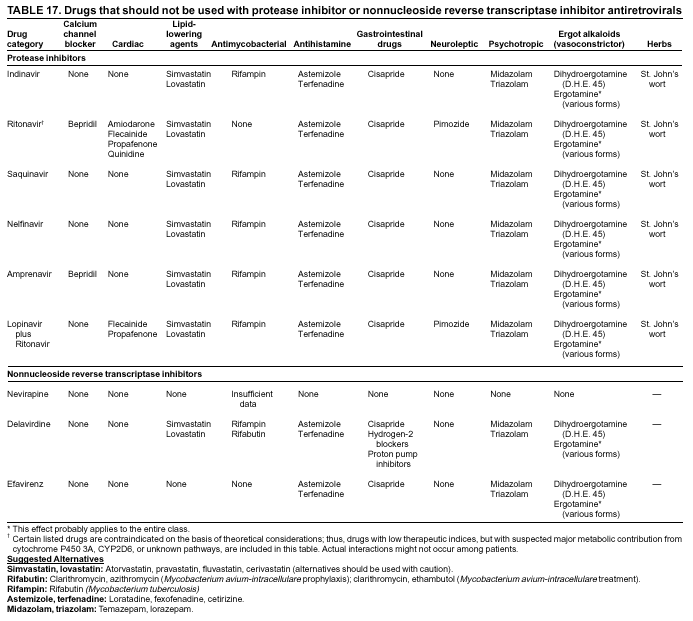 Return to top. Table 18 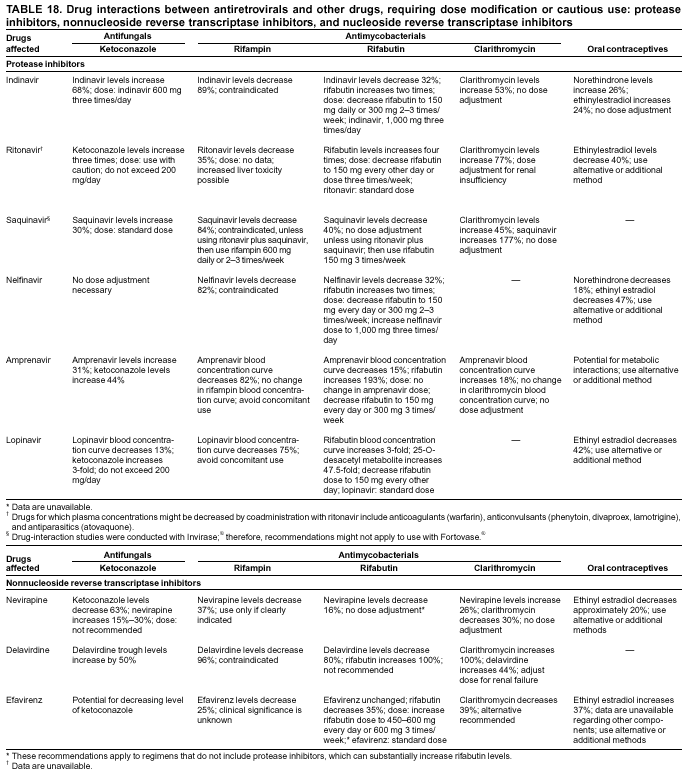  Return to top. Table 19 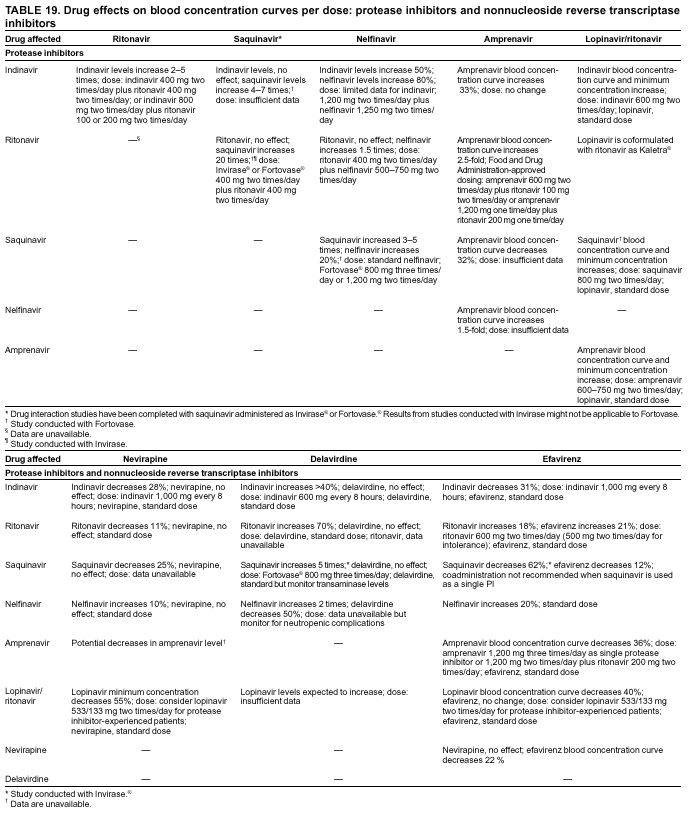 Return to top. Table 20 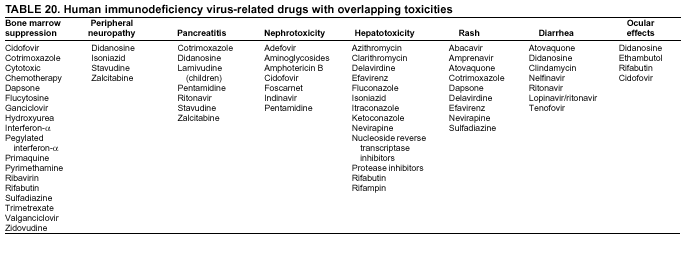 Return to top. Table 21  Return to top. Table 22  Return to top. Table 23  Return to top. Table 24 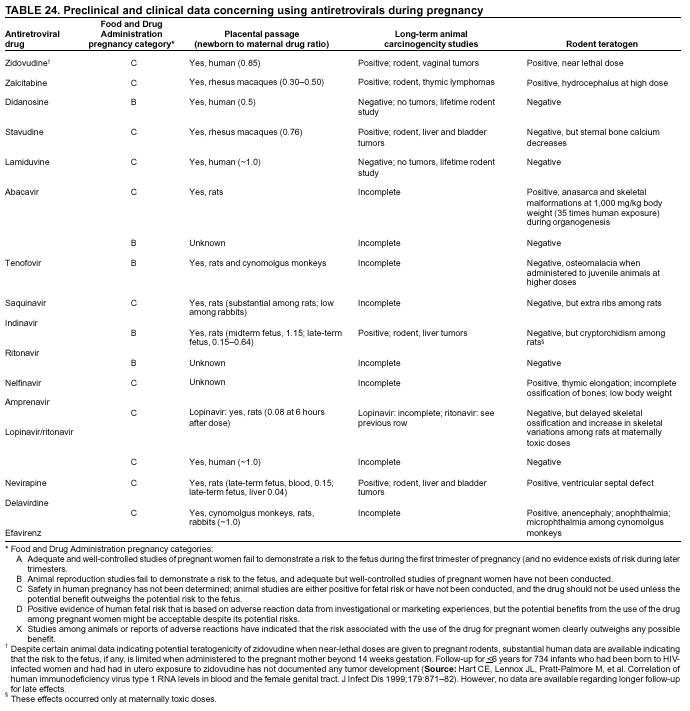 Return to top. List of Abbreviations Used in This Report
Panel on Clinical Practices for the Treatment of HIVConvened by the Department of Health and Human Services and the Henry J. Kaiser Family FoundationCo-Chairs: Anthony S. Fauci, M.D., National Institutes of Health, Bethesda, Maryland, and John G. Bartlett, M.D., Johns Hopkins University, Baltimore, Maryland. Co-Conveners: Eric P. Goosby, M.D., Department of Health and Human Services, Rockville, Maryland, and Jennifer Kates, M.A., M.P.A., Henry J. Kaiser Family Foundation, San Francisco, California. Executive Secretary: Mark Dybul, M.D., National Institutes of Health, Bethesda, Maryland. Assistant Executive Secretary: Alice K. Pau, Pharm.D., National Institutes of Health, Bethesda, Maryland. Members: Victoria Cargill, M.D., Oren J. Cohen, M.D., Henry Masur, M.D., Lynne M. Mofenson, M.D., and Douglas Brust, M.D., Ph.D.,* National Institutes of Health, Bethesda, Maryland; Jean Anderson, M.D., Johns Hopkins University School of Medicine, Baltimore, Maryland; Rodney C. Armstead, M.D., UHP Healthcare, Inc., Inglewood, California; A. Cornelius Baker, Whitman-Walker Clinic, Washington, D.C.; David Barr, J.D., George Washington University, Washington, D.C.; Samuel A. Bozzette, M.D., Ph.D., Veterans Affairs San Diego Healthcare System, San Diego, California; Charles C. J. Carpenter, M.D., Brown University School of Medicine, Providence, Rhode Island; Martin Delaney, Project Inform, San Francisco, California; Lawrence Deyton, M.D., Department of Veterans Affairs, Washington, D.C.; Wafaa El-Sadr, M.D., Harlem Hospital, New York, New York; Courtney Fletcher, Pharm.D., University of Colorado, Denver, Colorado; Gregg Gonsalves, Gay Men’s Health Crisis, New York, New York; Fred Gordin, M.D., Department of Veterans Affairs Medical Center, Washington, D.C.; Mark Harrington, Treatment Action Group, New York, New York; Martin S. Hirsch, M.D., Massachusetts General Hospital, Boston, Massachusetts; John W. Mellors, M.D., University of Pittsburgh Medical Center, Pittsburgh, Pennsylvania; James D. Neaton, Ph.D., University of Minnesota, Minneapolis, Minnesota; Sallie M. Perryman, New York State Department of Health, New York, New York; John Phair, M.D., Northwestern University Medical School, Chicago, Illinois; Robert T. Schooley, M.D., University of Colorado Health Science Center, Denver, Colorado; Renslow Sherer, M.D., Cook County Hospital/Rush Medical College, Chicago, Illinois; Stephen A. Spector, M.D., University of California, San Diego, La Jolla, California; Sharilyn K. Stanley, M.D., Texas Department of Health, Austin, Texas; Paul Volberding, M.D., University of California, San Francisco, California; Suzanne Willard, M.S.N., MCP/Hahnemann University, Philadelphia, Pennsylvania; T. Randolph Graydon, M.S., Health Care Financing Administration, Baltimore, Maryland; Debra Birnkrant, M.D., Heidi Jolson, and Jeffrey S. Murray, M.D., Food and Drug Administration, Rockville, Maryland; Jonathan E. Kaplan, M.D., CDC, Atlanta, Georgia; Abe M. Macher, M.D. and Joseph O’Neill, M.D., Health Resources and Services Administration, Rockville, Maryland; Lucille C. Perez, M.D., Substance Abuse and Mental Health Services Administration, Rockville, Maryland; Gerry Bally, M.D.,* Health Canada, Ottawa, Ontario; Sophia Chang, M.D.,* Center for Quality Management in HIV Care, Palo Alto, California; Jeff P. Jacobs,* AIDS Action Council, Washington, D.C.; Richard Marlink, M.D.,* Harvard School of Public Health, Cambridge, Massachusetts; Celia J. Maxwell, M.D.,* Howard University Hospital, Washington, D.C.; Howard Minkoff, M.D.,* Maimonides Medical Center, Brooklyn, New York; James M. Oleske, M.D.,* University of Medicine and Dentistry of New Jersey, Newark, New Jersey; and Daniel Simpson,* Indian Health Service, Rockville, Maryland. * Nonvoting observer.
Disclaimer All MMWR HTML versions of articles are electronic conversions from ASCII text into HTML. This conversion may have resulted in character translation or format errors in the HTML version. Users should not rely on this HTML document, but are referred to the electronic PDF version and/or the original MMWR paper copy for the official text, figures, and tables. An original paper copy of this issue can be obtained from the Superintendent of Documents, U.S. Government Printing Office (GPO), Washington, DC 20402-9371; telephone: (202) 512-1800. Contact GPO for current prices. **Questions or messages regarding errors in formatting should be addressed to mmwrq@cdc.gov.Page converted: 5/13/2002 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
This page last reviewed 5/13/2002
|