 |
|
|
|
|
|
|
| ||||||||||
|
|
|
|
|
|
|
||||
| ||||||||||
|
|
|
|
|
Persons using assistive technology might not be able to fully access information in this file. For assistance, please send e-mail to: mmwrq@cdc.gov. Type 508 Accommodation and the title of the report in the subject line of e-mail. Guidelines for Laboratory Testing and Result Reporting of Antibody to Hepatitis C Virus*Prepared by The material in this report originated in the National Center for Infectious Diseases, James M. Hughes, M.D., Director, and the Division of Viral Hepatitis, Harold S. Margolis, M.D., Director. Summary Testing for the presence of antibody to hepatitis C virus (anti-HCV) is recommended for initially identifying persons with hepatitis C virus (HCV) infection (CDC. Recommendations for prevention and control of hepatitis C virus [HCV] infection and HCV-related chronic disease. MMWR 1998;47[No. RR-19]:1--33). Testing for anti-HCV should include use of an antibody screening assay, and for screening test-positive results, a more specific supplemental assay. Verifying the presence of anti-HCV minimizes unnecessary medical visits and psychological harm for persons who test falsely positive by screening assays and ensures that counseling, medical referral, and evaluation are targeted for patients serologically confirmed as having been infected with HCV. However, substantial variation in reflex supplemental testing practices exists among laboratories, and an anti-HCV--positive laboratory report does not uniformly represent a confirmed positive result. These guidelines expand recommendations for anti-HCV testing to include an option for reflex supplemental testing based on screening-test--positive signal-to-cut--off (s/co) ratios. Use of s/co ratios minimizes the amount of supplemental testing that needs to be performed while improving the reliability of reported test results. These guidelines were developed on the basis of available knowledge of CDC staff in consultation with representatives from the Food and Drug Administration and public health, hospital, and independent laboratories. Adoption of these guidelines by all public and private laboratories that perform in vitro diagnostic anti-HCV testing will improve the accuracy and utility of reported anti-HCV test results for counseling and medical evaluation of patients by health-care professionals and for surveillance by public health departments. IntroductionTests to detect antibody to hepatitis C virus (anti-HCV) were first licensed by the Food and Drug Administration (FDA) in 1990 (1). Since that time, new versions of these and other FDA-approved anti-HCV tests have been used widely for clinical diagnosis and screening of asymptomatic persons. Persons being tested for anti-HCV are entitled to accurate and correctly interpreted test results. CDC has recommended that a person be considered to have serologic evidence of HCV infection only after an anti-HCV screening-test--positive result has been verified by a more specific serologic test (e.g., the recombinant immunoblot assay [RIBA®; Chiron Corporation, Emeryville, California]) or a nucleic acid test (NAT) (2). This recommendation is consistent with testing practices for hepatitis B surface antigen and antibody to human immunodeficiency virus (HIV), for which laboratories routinely conduct more specific reflex testing before reporting a result as positive (1,3). However, for anti-HCV, the majority of laboratories report a positive result based on a positive screening test result only, and do not verify these results with more specific serologic or nucleic acid testing unless ordered by the requesting physician. Unfortunately, certain health-care professionals lack an understanding of the interpretation of anti-HCV screening test results, when more specific testing should be performed, and which tests should be considered for this purpose. In certain clinical settings, false-positive anti-HCV results are rare because the majority of persons being tested have evidence of liver disease and the sensitivity and specificity of the screening assays are high. However, among populations with a low (<10%) prevalence of HCV infection, false-positive results do occur (4--11). This is of concern when testing is performed on asymptomatic persons for whom no clinical information is available, when persons are being tested for HCV infection for the first time, and when testing is being used to determine the need for postexposure follow-up. Without knowledge of the origin of the test sample or clinical information concerning the person being tested, the accuracy of a screening-test--positive result for any given specimen cannot be determined. Multiple reasons exist regarding why laboratories do not perform reflex supplemental testing for anti-HCV, including lack of an established laboratory standard for such testing, lack of understanding regarding the performance and interpretation of the screening and supplemental HCV tests, and the high cost of the supplemental HCV tests. To facilitate practice of reflex supplemental testing, the recommended anti-HCV testing algorithm has been expanded to include an option that uses the signal-to-cut--off (s/co) ratios of screening-test--positive results to minimize the number of specimens that require supplemental testing and provide a result that has a high probability of reflecting the person's true antibody status. BackgroundAvailable Anti-HCV Screening AssaysFDA-licensed or approved anti-HCV screening test kits being used in the United States comprise three immunoassays; two enzyme immunoassays (EIA) (Abbott HCV EIA 2.0, Abbott Laboratories, Abbott Park, Illinois, and ORTHO® HCV Version 3.0 ELISA, Ortho-Clinical Diagnostics, Raritan, New Jersey) and one enhanced chemiluminescence immunoassay (CIA) (VITROS® Anti-HCV assay, Ortho-Clinical Diagnostics, Raritan, New Jersey). All of these immunoassays use HCV-encoded recombinant antigens. Available Supplemental TestsSupplemental tests include a serologic anti-HCV assay and NATs for HCV RNA. In the United States, the only FDA-licensed supplemental anti-HCV test is the strip immunoblot assay (Chiron RIBA® HCV 3.0 SIA, Chiron Corp., Emeryville, California). RIBA 3.0 uses both HCV-encoded recombinant antigens and synthetic peptides. Because it is a serologic assay, it can be performed on the same serum or plasma sample collected for the screening anti-HCV assay. FDA-approved diagnostic NATs for qualitative detection of HCV RNA using reverse transcriptase polymerase chain reaction (RT-PCR) amplification include AMPLICOR® Hepatitis C Virus (HCV) Test, version 2.0 and COBAS AMPLICOR® Hepatitis C Virus Test, version 2.0 (Roche Molecular Systems, Branchburg, New Jersey), which have a lower limit of detection of approximately 50 IU/mL (12). Detection of HCV RNA by these tests requires that the serum or plasma sample be collected and handled in a manner suitable for NAT and that testing be performed in a laboratory with facilities established for this purpose (see Recommendations). Other NATs for HCV RNA, both qualitative and quantitative, are available on a research-use basis from multiple manufacturers of diagnostic reagents, and certain laboratories perform NATs by using in-house laboratory methods and reagents (12,13). Interpreting Anti-HCV Test ResultsScreening Immunoassay Test Results Anti-HCV testing includes initial screening with an immunoassay. Criteria for interpretation of a reactive† anti-HCV immunoassay result are based on data from clinical studies performed under the auspices of each manufacturer. For EIAs (e.g., HCV EIA 2.0 and HCV Version 3.0 ELISA), specimens with a reactive result are retested in duplicate. If the result of either duplicate test is reactive, the specimen is defined as repeatedly reactive and is interpreted as screening-test--positive. For CIAs, (e.g., VITROS Anti-HCV assay), specimens with a single reactive result are considered screening-test--positive and do not require retesting. The specificity of the HCV EIA 2.0 and HCV Version 3.0 ELISA is >99%. However, among a population with a low prevalence of infection, even a specificity of 99% does not provide the desired predictive value for a positive test. Among immunocompetent populations with anti-HCV prevalences <10% (e.g., volunteer blood donors, active duty and retired military personnel, persons in the general population, health-care workers, or clients attending sexually transmitted disease [STD] clinics), the proportion of false-positive results with HCV EIA 2.0 or HCV Version 3.0 ELISA averages approximately 35% (range: 15%--60%) (4--11; CDC, unpublished data, 2002). Among immunocompromised populations (e.g., hemodialysis patients), the proportion of false-positive results averages approximately 15% (14; CDC, unpublished data, 2002). For this reason, not relying exclusively on anti-HCV screening-test--positive results to determine whether a person has been infected with HCV is critical. Rather, screening-test--positive results should be verified with an independent supplemental test with high specificity. Supplemental Serologic Test Results The strip immunoblot assay (RIBA), a supplemental anti-HCV test with high specificity, is performed on screening-test--positive samples and provides results that are interpreted as positive, negative, or indeterminate. A positive RIBA result is interpreted as anti-HCV--positive (Box). Although the presence of anti-HCV does not distinguish between current or past infection, a confirmed anti-HCV--positive result indicates the need for counseling and medical evaluation for HCV infection, including additional testing for the presence of virus (NAT for HCV RNA) and liver disease (e.g., alanine aminotransferase [ALT]) (2,15). Anti-HCV testing usually does not need to be repeated after the anti-HCV--positive result has been confirmed. A negative RIBA result is interpreted as anti-HCV--negative and indicates a false-positive screening test result. In this situation, the additional testing with RIBA minimizes unnecessary medical visits and psychological harm from reporting a false-positive screening test result. Typically, persons whose anti-HCV test results are negative (screening-test--negative or RIBA-negative) are considered uninfected (Box). However, false-negative anti-HCV test results can occur during the first weeks after infection (i.e., before antibody is detectable or during seroconversion), although HCV RNA can be detected as early as 1--2 weeks after exposure to the virus (16,17). Rarely, antibody seroconversion might be delayed for months after exposure (18,19). In certain persons whose HCV infection has resolved, anti-HCV declines below detectable levels (20). Occasionally, persons with chronic HCV infection, including those who are immunocompromised, are persistently anti-HCV--negative, and detection of HCV RNA might be the only evidence of infection (14,21). An indeterminate RIBA result indicates that the anti-HCV result cannot be determined (Box). Indeterminate anti-HCV supplemental test results have been observed in recently infected persons who are in the process of seroconversion, and occasionally in persons chronically infected with HCV (22). Indeterminate results also might indicate a false-positive screening test result, which is the most common interpretation for these results among those at low risk for HCV infection (23,24). Another sample should be collected for repeat anti-HCV testing (>1 month later) or for HCV RNA testing. Supplemental NAT Results NATs that detect HCV RNA also can be used as supplemental tests for anti-HCV. They are used commonly in clinical practice for diagnosis of acute and chronic HCV infection and for evaluating and managing patients with chronic hepatitis C. If the NAT result is positive in persons with a positive screening test result, NAT has the advantage of detecting the presence of active HCV infection as well as verifying the presence of anti-HCV (Box). If the NAT result is negative in persons with a positive screening test result, the HCV antibody or infection status cannot be determined. Among persons with these results, additional testing with RIBA is necessary to verify the anti-HCV result and determine the need for counseling and medical evaluation (Box); if the anti-HCV screening test results are judged falsely positive (i.e., RIBA-negative), no further evaluation of the person is needed; whereas if the anti-HCV screening test results are verified as positive by RIBA, the person should undergo medical evaluation, including serial determinations of HCV RNA and ALT activity. Certain situations exist in which the HCV RNA result can be negative in persons with active HCV infection. As the titer of anti-HCV increases during acute infection, the titer of HCV RNA declines (17). Thus, HCV RNA is not detectable in certain persons during the acute phase of their hepatitis C, but this finding can be transient and chronic infection can develop (25). In addition, intermittent HCV RNA positivity has been observed among persons with chronic HCV infection (21,26,27). Therefore, in the absence of additional clinical information, the significance of a single negative HCV RNA result is unknown, and the need for further medical evaluation is determined by verifying anti-HCV status. A negative HCV RNA result also can indicate resolved infection. Among anti-HCV--positive persons who acquired their HCV infection as older adults (aged >45 years), 15%--25% apparently resolve their infection; this proportion is higher (40%--45%) among anti-HCV--positive persons who acquired their infection as children or younger adults (20). To determine if HCV infection has resolved, a negative HCV RNA result should be demonstrated on multiple occasions; however, such follow-up testing is indicated only in persons with serologically confirmed anti-HCV positive results. Anti-HCV Testing PracticesMultiple commercial, hospital-based, and public health laboratories that perform anti-HCV testing routinely report screening test results only. More specific testing (i.e., RIBA or NAT) is performed only when ordered by a physician. Moreover, in certain laboratories, more specific tests are not available. During 2002, two surveys regarding anti-HCV testing practices were performed, one by the Association of Public Health Laboratories (Barbara Werner, Ph.D., Association of Public Health Laboratories, personal communication, September 2002) and one by a Veterans Affairs (VA) Medical Center (D. Robert Dufour, M.D., VA Medical Center, Washington, D.C., personal communication, October 2002). Forty-three (80%) of 54 U.S. state and territorial public health laboratories and 67 (39%) of 172 VA medical center laboratories responded (Table 1). Of the respondents, the public health laboratories were less likely to offer screening or supplemental tests for HCV than were the hospital-based VA laboratories. However, the public health laboratories that did offer both types of testing were more likely to perform reflex supplemental testing than were the hospital-based laboratories, 75% of which performed supplemental testing only by physician request. Regarding the type of supplemental testing performed, the majority of hospital-based laboratories performed only NATs, whereas the public health laboratories most commonly performed either RIBA alone or NAT followed by RIBA if the NAT result was negative. Although substantial differences existed in testing practices between and among these two types of laboratories, the majority of public and private sector laboratories depend on the requesting physician to be knowledgeable concerning the appropriate tests to order and the correct interpretation of their results. However, a general lack of understanding exists among health-care professionals regarding the interpretation of screening test results, when more specific testing should be performed, and which tests should be considered for this purpose. Using Screening-Test--Positive S/Co Ratios To Determine Need for Reflex Supplemental TestingAnalysis of early versions of anti-HCV EIA results from volunteer blood donors indicated that average repeatedly reactive s/co ratios could be used to predict supplemental test-positive results (28). Similar data from volunteer blood donors were generated by using HCV Version 3.0 ELISA, for which the average s/co ratios of 24,700 samples repeatedly reactive for anti-HCV were compared with their RIBA 3.0 results (Susan Stramer, Ph.D., American Red Cross, personal communication, March 1999). Overall, 64.0% were RIBA-positive. The proportion that tested RIBA-positive was 5.8% for samples with an average s/co ratio 1.0--2.9; 37.1% for those with average s/co ratio 3.0--3.4; 67% for those with average s/co ratio 3.5--3.7; 88.1% for those with average s/co ratio 3.8--3.9; and 94.1% for those with average s/co ratio >4.0. Additional data from other populations were generated by CDC to determine if a specific s/co ratio could be identified that would predict a true antibody-positive result >95% of the time, regardless of the anti-HCV prevalence or characteristics of the population being tested. The anti-HCV screening tests evaluated were the two FDA-licensed EIAs, HCV EIA 2.0 and HCV Version 3.0 ELISA, and the one FDA-approved CIA, VITROS Anti-HCV assay. EIAs All specimens with EIA screening-test--positive results were tested by RIBA 3.0, and a sample of screening-test--positive specimens were tested for HCV RNA by >2 of the following NAT methods: transcription-mediated amplification (TMA) (Procleix™, Chiron Corporation, Emeryville, California); AMPLICOR; and nested RT-PCR (13). Test results were used from serum samples that had been collected as part of CDC-sponsored anti-HCV seroprevalence studies that were conducted among different groups of asymptomatic persons (Robert Gunn, M.D., San Diego County Department of Health and Human Services Agency; Steven Harris, M.D., Travis County, Texas Department of Health; Lu-Yu Hwang, M.D., University of Texas --- Houston School of Public Health; Leslie Tobler, Ph.D., Blood Centers of the Pacific, San Francisco; Gayle Shimokura, University of North Carolina at Chapel Hill School of Public Health; Isaac Weisfuse, M.D., New York City Department of Health and Mental Hygiene, personal communications, 2001--2002; CDC, unpublished data, 2002). Anti-HCV prevalences ranged from 0.8% to 25% (Table 2). The proportion of screening-test--positive results that were serologically confirmed as anti-HCV--positive (i.e., RIBA-positive) increased as the anti-HCV prevalence in the population increased (Table 2). Conversely, the proportion of screening-test--positive results that were falsely antibody-positive (RIBA-negative) or RIBA-indeterminate was inversely related to prevalence (Table 2). For each study group, the proportion of screening-test--positive results that were RIBA-positive increased as the screening-test--positive average s/co ratios increased (Figure 1). On the basis of these data, screening-test--positive average s/co ratios >3.8 were highly predictive of RIBA positivity (>95%), with limited variability (95%--97%) between groups with different prevalences (Table 2). Screening-test--positive average s/co ratios >3.8 also were highly predictive of HCV RNA positivity, although the proportions that were HCV RNA-positive were slightly lower than those for RIBA (Table 2). These results indicate that for licensed EIAs, reporting anti-HCV screening-test--positive results as anti-HCV positive for samples with average s/co ratios >3.8 would be highly predictive of the true anti-HCV status. Reflex supplemental testing before reporting the anti-HCV results could be limited to screening-test--positive samples with average s/co ratios <3.8. The feasibility of this approach is supported further by the limited proportion (2.4%) of samples from persons at high risk that have s/co ratios below this cut-off value. When testing for anti-HCV is performed on persons at increased risk for infection as recommended (2), a limited number of samples will require additional testing. CIA The relation between s/co ratios and RIBA 3.0 results also was evaluated for specimens that were screening-test--positive by CIA (i.e., reactive by VITROS Anti-HCV) from four groups. These included a group of 162 volunteer blood donors with substantially low anti-HCV prevalence (Leslie Tobler, Ph.D., Blood Centers of the Pacific, San Francisco, personal communication, September 2002), a group of 163 persons with low anti-HCV prevalence (college students, persons in the general population, and health-care workers as described previously), a group of 219 hemodialysis patients with intermediate anti-HCV prevalence (as described previously), and a group of 689 hospital-based patients with high anti-HCV prevalence (signs or symptoms of liver disease or risk factors for HCV infection) (D. Robert Dufour, M.D., VA Medical Center, Washington, D.C., and Michael De Lucia, Ortho-Clinical Diagnostics, personal communications, September 2002). Overall, the proportion of CIA screening-test--positive samples that tested RIBA-positive was 77.8% among the blood donors, 74.2% among the low prevalence group, 86.3% among the hemodialysis patients, and 94.5% among the high prevalence group. The direct relation between increasing s/co ratios and RIBA positivity that was observed among samples tested with the two EIAs evaluated by CDC also was observed among the samples tested with the CIA (Figure 2). However, the range of screening-test--positive s/co ratios obtained with VITROS Anti-HCV was greater than that obtained with HCV EIA 2.0 or HCV Version 3.0 ELISA; thus, s/co ratios that were highly predictive of RIBA positivity also were higher. Using VITROS Anti-HCV, an s/co ratio of >8 predicted RIBA positivity in 95%--98% of the screening-test--positive samples (Figure 2). The proportion of CIA samples with low s/co ratios was inversely related to anti-HCV prevalence (i.e., 4.9% in the high prevalence group, 8.7% in the intermediate prevalence group, and 21.5% in the low prevalence group). These results indicate that for the FDA-approved CIA, reflex supplemental testing of screening-test--positive samples also could be limited to those with a low (<8) s/co ratio; and among persons at increased risk for infection, <5% will have s/co ratios below the cut-off value. Estimated Costs of Implementing Reflex Supplemental Testing Based on Screening-Test--Positive S/Co RatiosTo assist laboratories in assessing the potential financial impact of implementing reflex supplemental testing for screening-test--positive samples with low s/co ratios, the incremental costs associated with such testing were estimated for three hypothetical populations of 10,000 persons each, representing anti-HCV prevalences of 2%, 10%, and 25%, respectively (similar to those of the groups evaluated previously). For each population, the costs of performing the screening test (by using EIAs as the example) and each of two different supplemental testing schemes (schemes 1 and 2) were compared with the cost of performing only the screening test (base scheme). All schemes included performing a screening EIA on each sample and repeating initially reactive specimens in duplicate. Scheme 1 also included RIBA testing on all screening-test--positive samples with average s/co ratios <3.8, and scheme 2 included NAT testing on all screening-test--positive samples with average s/co ratios <3.8, followed by RIBA on those that were NAT-negative. The increased costs for schemes 1 and 2 were calculated per sample tested compared with the base scheme. For RIBA and NAT, minimum and maximum costs were estimated; minimum costs were defined as costs for reagents only, and maximum costs were defined as costs incurred for tests performed by a referral laboratory. The following assumptions were made:
Compared with performing only the screening test, performing reflex RIBA testing on all screening-test--positive samples with average s/co ratios <3.8 (scheme 1) increases the cost of testing per sample for immunocompetent populations from a minimum of 5%--12% ($0.41--$0.66) to a maximum of 13%--30% ($1.00--$1.60), depending on the anti-HCV prevalence of the population being tested (Figure 3). For hemodialysis patients, the cost increases from a minimum of 16% ($1.00) to a maximum of 38% ($2.44). Performing reflex NATs on all screening-test--positive samples with average s/co ratios <3.8, followed by RIBA on those that are NAT-negative (scheme 2), increases the cost of testing per sample for immunocompetent populations from a minimum of 9%--21% ($0.73--$1.14) to a maximum of 37%--85% ($2.88--$4.54), compared with performing only the screening test. For hemodialysis patients, the cost increases from a minimum of 27% ($1.73) to a maximum of 109% ($6.88). The higher incremental costs of scheme 2 compared with scheme 1 are because virtually all the screening-test--positive samples with s/co ratios <3.8 test HCV RNA-negative and require follow-up testing with RIBA to verify anti-HCV status. RecommendationsRationaleTesting for HCV infection by using anti-HCV is performed for 1) clinical diagnosis of patients with signs or symptoms of liver disease; 2) management of occupational and perinatal exposures; and 3) screening asymptomatic persons to identify HCV-infected persons who should receive counseling and medical evaluation. Anti-HCV test results also are used for public health surveillance to monitor incidence and prevalence and to target and evaluate HCV prevention efforts. Anti-HCV testing is performed in multiple settings, including hospitals and other health-care facilities, physicians' offices, health department clinics, HIV or other freestanding counseling and testing sites, employment sites, and health fairs. The interpretation of anti-HCV screening-test--positive results in these settings can be problematic. Clinical information related to the persons tested often is lacking, and even persons with risk factors for HCV infection might be at sufficiently low enough risk for infection that their screening test results could be falsely positive (e.g., health-care professionals are at occupational risk for HCV infection, but their overall prevalence of infection is low) (29). Without knowledge of the origin of the test sample or clinical information related to the person being tested, the accuracy of a screening-test--positive result for any given specimen cannot be determined. However, despite previous recommendations for reflex supplemental testing of all anti-HCV screening-test--positive results (2), the majority of laboratories report positive anti-HCV results based only on a positive screening assay. To facilitate and improve the practice of reflex supplemental testing, the recommended anti-HCV testing algorithm has been expanded to include an option for more specific testing based on the s/co ratios of screening-test--positive results that can be implemented without substantial increases in testing costs. Implementation of these recommendations will provide more reliable results for physicians and their patients, so that further counseling and clinical evaluation are limited to those confirmed to have been infected with HCV. This is critical for persons being tested for HCV infection for the first time, for persons being tested in nonclinical settings, and for those being tested to determine the need for postexposure follow-up. Implementation of these recommendations also will improve public health surveillance systems for monitoring the effect of HCV prevention and control activities. Laboratory Algorithm for Anti-HCV Testing and Result ReportingAll laboratories that provide anti-HCV testing should perform initial screening with an FDA-licensed or approved anti-HCV test according to the manufacturer's labeling.
Reflex Supplemental Testing Based on Screening-Test--Positive S/Co Ratios
Reflex Supplemental Testing on All Specimens with Screening-Test--Positive Results
Considerations When Choosing a Reflex Supplemental Testing Option Serologic Supplemental Testing.
Nucleic Acid Supplemental Testing.
Other Reflex Supplemental Testing Options Certain laboratories might choose to modify the recommended supplemental testing options to provide additional information before reporting results. One such modification might include reflex NAT of screening-test--positive results with high s/co ratios, which might be of interest to hospital-based laboratories that usually test specimens from patients being evaluated for liver disease. If the NAT result is positive, the presence of active HCV infection can be reported as well as a positive anti-HCV result. However, if the NAT result is negative, reflex RIBA testing still is required before reporting the results to verify the anti-HCV status. Certain specimens will test RIBA-positive, indicating that the person should receive further evaluation, including repeat testing for HCV RNA (see Interpretation of Anti-HCV Test Results). ImplementationTo implement these recommendations for anti-HCV testing and result reporting, laboratories should review their present testing and reporting methods and determine how those should be modified. This process should include
Laboratories that select a reflex supplemental testing option based on screening-test--positive s/co ratios need to ensure that their analyzers generate optical density (OD) values in a range sufficient to calculate s/co ratios at or above the value defined as a high s/co ratio for the screening test being used. The s/co ratio is calculated by dividing the OD value of the sample being tested by the OD value of the assay cut-off for that run. Depending on the type of equipment in the laboratory, the calculation of s/co ratios might be automatically performed by the analyzer or require that the technician manually perform the calculation. For screening tests that require only one reactive result to indicate a screening-test--positive result (e.g., VITROS Anti-HCV), the s/co ratio of the reactive result is used to determine the next step in the algorithm (i.e., reporting the result or reflex supplemental testing). For screening tests that require repeating initially reactive results in duplicate (e.g, HCV EIA 2.0 and HCV Version 3.0 ELISA), the s/co ratio of each of the duplicate results is calculated. The average of the s/co ratios of the reactive results is used to determine the next step in the algorithm. If all three results are reactive for the sample, the average s/co ratio can be determined either by averaging the ratios of all three or by averaging only the ratios of the two duplicate reactive results. If only one of the duplicate results is reactive, the average s/co ratio is determined by averaging the ratios from the initial reactive result and the one duplicate reactive result. For those screening-test--positive samples that undergo reflex supplemental testing (according to the testing option chosen), the screening test anti-HCV results should not be reported before the results from the additional testing are available. If necessary, an interim report can be issued indicating that the result is pending. This procedure should be followed even if the laboratory does not perform the supplemental testing in-house, but sends the sample to another reference laboratory for such testing. After the results are received from the reference laboratory, the final results can be reported on the basis of the testing performed by both laboratories. The reported results should be accompanied by interpretive comments as determined by each laboratory (Table 3). The content of these comments will vary on the basis of type of supplemental testing option selected by the laboratory. These comments are critical if screening-test--positive results are reported as anti-HCV--positive on the basis of high s/co ratios, because the health-care professional or other person interpreting the results needs to understand the limitations of the testing option used. Before implementation, the laboratory staff should be educated regarding new methods of testing, calculating, and reporting final results for the selected testing option. Laboratories also should inform and educate all customers regarding the planned changes and what effects they will have on test results generated. This information should be disseminated as widely as possible (e.g., by laboratory bulletins, letters, Internet, or continuing education programs). Depending on the setting, reimbursement of clinical laboratory tests used for reflex supplemental testing might depend on documentation that the physician ordered the tests. This documentation can be achieved through a printed requisition form that clearly identifies for anti-HCV the specified level of results of the screening test that will trigger additional supplemental testing and what type(s) of supplemental testing will be performed. In addition, each of the supplemental tests (e.g., RIBA or NAT) that are offered by the laboratory should be listed separately, because physicians should be able to order these as they deem necessary for further medical evaluation. Future ConsiderationsAs new anti-HCV screening assays are approved or licensed for use, each will need to be evaluated for its specificity among populations with different anti-HCV prevalences. In addition, before using a new assay to perform reflex supplemental testing based on screening-test--positive s/co ratios, the s/co ratio value at or above which supplemental test results are positive >95% of the time in populations in which the test will be used, should be determined. Such documentation also should be required for approved screening assays if any modifications are made to the testing procedures that might affect the s/co ratio values. Similarly, the relation between screening-test--positive results and the results of newly available supplemental tests will need to be evaluated. Acknowledgments We acknowledge Garth Austin, M.D., Ph.D., Veterans Affairs Medical Center, Atlanta, Georgia, for performing serologic testing, and D. Robert Dufour, M.D., Veterans Affairs Medical Center, Washington, D.C., Leslie Tobler, Ph.D., and Michael Busch, M.D., Ph.D., Blood Centers of the Pacific, San Francisco, California, and Susan Stramer, Ph.D., American Red Cross, Gaithersburg, Maryland, for sharing their data and expertise and for performing serologic and nucleic acid testing to generate additional information that was needed to develop these guidelines. References
Terms and Abbreviations Used in This Report
* These guidelines are not intended to be used for blood, plasma, organ, tissue, or other donor screening or notification as provided for under FDA guidance or applicable regulations. They also are not intended to change the manufacturer's labeling for performing a specific test. † The terms reactive or nonreactive are used to describe serum or plasma specimen test results from anti-HCV screening immunoassays before final interpretation. The terms positive and negative are used to describe the final interpretation of screening immunoassay test results (e.g., screening-test--positive indicates that the specimen tested is repeatedly reactive by EIA or reactive by CIA, or screening-test--negative indicates that the specimen tested is nonreactive or not repeatedly reactive). The terms positive, indeterminate, and negative are used to describe the interpretation of RIBA results based on reactivity with a specific pattern of bands. § Data are available from three screening assays. For the two EIAs (HCV EIA 2.0 or HCV Version 3.0 ELISA), high s/co ratios are defined as screening-test--positive results with average s/co ratios >3.8, and low s/co ratios as screening-test--positive results with average s/co ratios <3.8. For CIA (VITROS Anti-HCV), high s/co ratios are defined as screening-test--positive results with s/co ratios >8, and low s/co ratios as screening-test--positive results with s/co ratios <8. Laboratory Testing and Result Reporting of Antibody to Hepatitis C Virus Working Group Members Miriam J. Alter, Ph.D., Wendi L. Kuhnert, Ph.D., Lyn Finelli, Dr.P.H., Division of Viral Hepatitis, National Center for Infectious Diseases, CDC; William Schalla, M.S., Ana Stankovic, M.D., Ph.D., Robert Martin, Dr.P.H., Division of Laboratory Systems, Public Health Program Practice Office, CDC; Robin Biswas, M.D., Brian Harvey, M.D., Ph.D., Food and Drug Administration, Bethesda, Maryland; John Bryan, M.D., American Society for Clinical Pathology, Chicago, Illinois; D. Robert Dufour, M.D., National Academy of Clinical Biochemistry, Washington, D.C.; Mark Jandreski, Ph.D., American Association for Clinical Chemistry, Washington, D.C.; Amy Leber, Ph.D., American Clinical Laboratory Association, Washington, D.C.; Frederick Nolte, Ph.D., Emory University School of Medicine, Atlanta, Georgia; Robin Stombler, American Society for Clinical Pathology, Washington, D.C.; David Sundwall, M.D., American Clinical Laboratory Association, Washington, D.C.; James Versalovic, M.D., Ph.D., College of American Pathologists, Northfield, Illinois; Judith Wethers, M.S., New York State Public Health Laboratory, Albany, New York; Barbara Werner, Ph.D., Association of Public Health Laboratories, Washington, D.C.; and Susanne Zanto, Association of Public Health Laboratories, Washington, D.C. Table 1 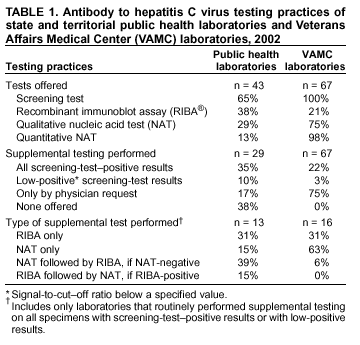 Return to top. Figure 1 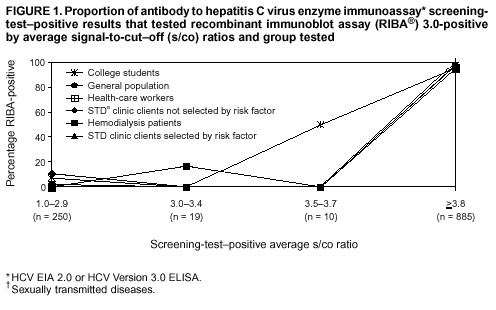 Return to top. Table 2 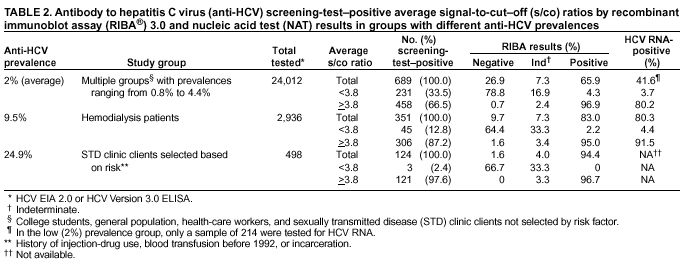 Return to top. Figure 2 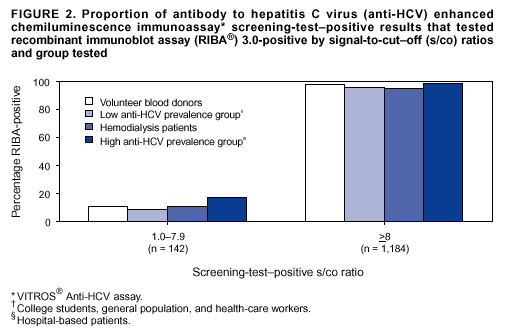 Return to top. Table 3 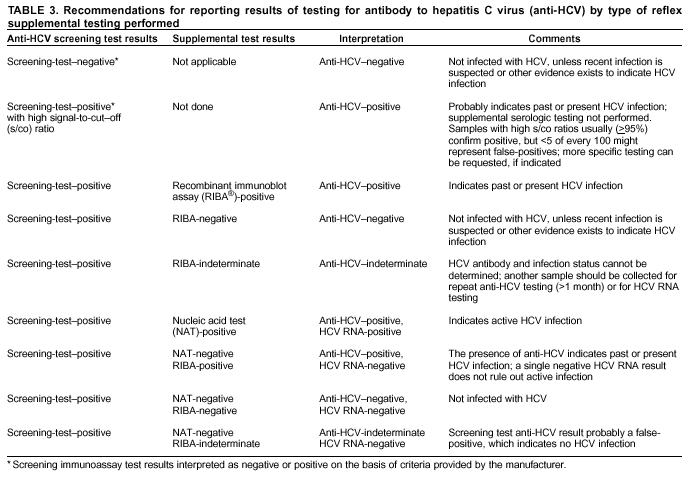 Return to top. Figure 3 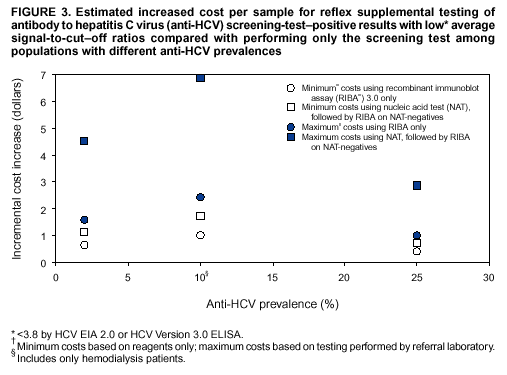 Return to top. Figure 4 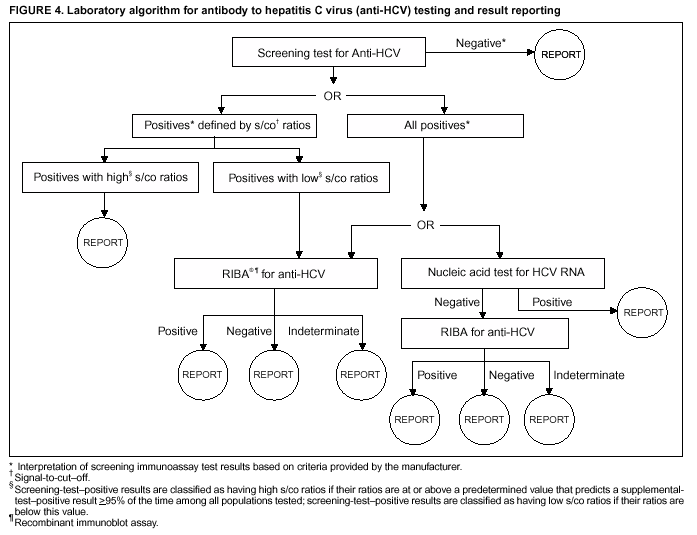 Return to top. Box  Return to top.
Disclaimer All MMWR HTML versions of articles are electronic conversions from ASCII text into HTML. This conversion may have resulted in character translation or format errors in the HTML version. Users should not rely on this HTML document, but are referred to the electronic PDF version and/or the original MMWR paper copy for the official text, figures, and tables. An original paper copy of this issue can be obtained from the Superintendent of Documents, U.S. Government Printing Office (GPO), Washington, DC 20402-9371; telephone: (202) 512-1800. Contact GPO for current prices. **Questions or messages regarding errors in formatting should be addressed to mmwrq@cdc.gov.Page converted: 1/27/2003 |
|||||||||||||||||||||||||||||||||||||||||||||||
This page last reviewed 1/27/2003
|