|
|
|
|
|
|
|
| ||||||||||
|
|
|
|
|
|
|
||||
| ||||||||||
|
|
|
|
|
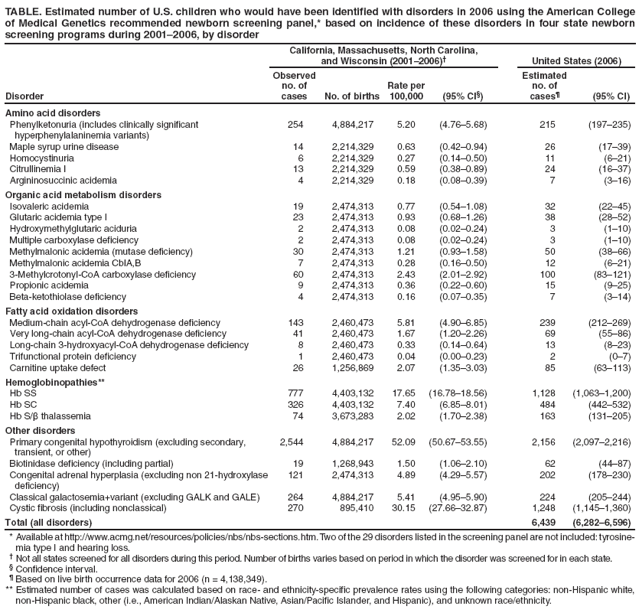
Persons using assistive technology might not be able to fully access information in this file. For assistance, please send e-mail to: mmwrq@cdc.gov. Type 508 Accommodation and the title of the report in the subject line of e-mail. Impact of Expanded Newborn Screening --- United States, 2006Universal newborn screening for selected metabolic, endocrine, hematologic, and functional disorders is a well-established practice of state public health programs. Recent developments in tandem mass spectrometry (MS/MS), which is now capable of multi-analyte analysis in a high throughput capacity, has enabled newborn screening to include many more disorders detectable from a newborn blood spot (1). In 2006, to address the substantial variation that existed from state to state in the number of disorders included in newborn screening panels, the American College of Medical Genetics (ACMG), under guidance from the Health Resources and Services Administration, recommended a uniform panel of 29 disorders, which was subsequently endorsed by the federal Advisory Committee on Heritable Disorders in Newborns and Children (2). After 2006, most states began to expand their panels to include all 29 disorders; currently, 21 states and the District of Columbia have fully implemented the ACMG panel.* To estimate the burden to state newborn screening programs resulting from this expansion (3), CDC used 2001--2006 data from those states with well-established MS/MS screening programs† to estimate the number of children in the United States who would have been identified with disorders in 2006 if all 50 states and the District of Columbia had been using the ACMG panel. This report describes the results of that analysis, which indicated that, although such an expansion would have increased the number of children identified by only 32% (from 4,370 to 6,439), these children would have had many rare disorders that require local or regional capacity to deliver expertise in screening, diagnosis, and management. The findings underscore the need for public health and health-care delivery systems to build or expand the programs required to manage the rare disorders detected through expanded newborn screening, while also continuing programs to address more common disorders. Data on the number of children detected by newborn screening with a confirmed disorder were obtained from four state health departments: California, Massachusetts, North Carolina, and Wisconsin. Massachusetts, North Carolina, and Wisconsin were included because they were the only states with well-established MS/MS screening programs that had been screening for a majority of the disorders in the ACMG panel for at least 6 years. California was included because the state implemented a more recent expansion (in 2005) after a well-documented pilot study (4) and had a large number of births per year. The 2006 estimates were based on newborn screening data for the period 2001--2006; not all states contributed data for all the disorders for the entire period. Tyrosinemia type 1 and hearing loss, two disorders included in the ACMG panel, were not included in this analysis. Tyrosinemia type 1 was not included based on recent evidence that current laboratory screening methods are insufficient for detecting the majority of cases of this disorder (5). Data for children identified from newborn hearing screening were not included because they are reported in separate systems at the state level. The cumulative incidence for each disorder was determined by summing the total number of cases observed in all four state newborn screening programs for the periods that each state was screening for the disorder, and dividing that sum by the total number of live births in the four states combined during the respective periods. To estimate the number of live births in the United States affected by each disorder in 2006, CDC multiplied the disorder-specific rate by the number of live births in the United States in 2006. All live birth data (state and U.S.) were obtained from National Center for Health Statistics vital records files using state of occurrence of the live birth rather than state of residence, which is analogous to the newborn screening procedures at the state level. For hemoglobinopathies, the estimate of live births affected was based on race- and ethnicity-specific prevalence rates using the following categories: non-Hispanic white, non-Hispanic black, other (i.e., American Indian/Alaskan Native, Asian/Pacific Islander, and Hispanic), and unknown race/ethnicity. Exact Poisson 95% confidence intervals were calculated for the disorder-specific rates that were used to estimate the upper and lower bounds for the estimated number of cases in the United States for 2006 (6). Because of the large number of total cases, the 95% confidence interval for this value was estimated using the normal approximation. The estimated number of cases of disorders that would have been identified in 2006 using the ACMG panel was 6,439, 32% more than the 4,370 that would have been identified otherwise (Table). The three hemoglobinopathies and congenital hypothyroidism combined accounted for 61% of the total estimated number of cases. Ten disorders accounted for an estimated 100 or more cases (phenylketonuria, 3-methylcrotonyl-CoA carboxylase deficiency, medium-chain acyl-CoA dehydrogenase deficiency, Hb SS, Hb SC, Hb S/β thalassemia, congenital hypothyroidism, congenital adrenal hyperplasia, galactosemia, and cystic fibrosis). Four disorders accounted for an estimated 50 or more cases (methylmalonic acidemia attributed to mutase deficiency, very long-chain acyl-CoA dehydrogenase deficiency, carnitine uptake defect, and biotinidase deficiency). Nine of the disorders accounted for an estimated 15 or fewer cases. Reported by: B Therrell, National Newborn Screening and Genetics Resource Center, Austin, Texas. F Lorey, Genetic Diseases Laboratory, California Dept of Health Svcs. R Eaton, Univ of Massachusetts Medical School, Boston, Massachusetts. D Frazier, Div of Genetics and Metabolism, Univ of North Carolina at Chapel Hill. G Hoffman, Wisconsin State Laboratory of Hygiene. C Boyle, D Green, Div of Birth Defects and Developmental Disabilities, O Devine, National Center for Birth Defects and Developmental Disabilities; H Hannon, Div of Laboratory Sciences, National Center for Environmental Health, CDC. Editorial Note:With advances in treatment and the ACMG-recommended expansion of the newborn screening panel, the adverse health consequences of various disorders can now be minimized or avoided. For example, one of the disorders included in the expanded panel, medium-chain acyl-CoA dehydrogenase deficiency (MCAD), involves a simple treatment (i.e., avoiding fasting); thus, proper medical management of a child identified with MCAD can be lifesaving. The findings in this report indicate that if all state newborn screening programs had used the expanded ACMG panel in 2006 to screen for disorders identifiable from a newborn blood spot, 6,439 newborns would have been identified with one of these disorders. In 2003, before the recommendation to expand the screening panel, all but four states were screening for six disorders (galactosemia, hemoglobinopathies [Hb SS, Hb SC, and Hb S/β thalassemia], phenylketonuria, and congenital hypothyroidism).§ These six disorders represent 68% of the estimated caseload for 2006, and congenital hypothyroidism and the hemoglobinopathies account for the majority of these cases. The addition of 21 disorders, many of which were estimated to have fewer than 15 cases, underscores the dual challenge of continuing the screening program for the more common disorders while also building the expertise and resources to manage the many rare disorders. Several states are addressing these resource concerns by outsourcing laboratory testing and working collaboratively to share expertise on laboratory, diagnostic, and treatment issues. At the federal level, the Health Resources and Services Administration is facilitating development of technical and clinical expertise through a regional network of technical centers; in addition, a federal advisory committee, the Committee on Heritable Disorders in Newborns and Children, provides guidance on the appropriate application of newborn screening tests, technologies, policies, and guidelines (7). The continued success of the expanded screening programs depends on the development of surveillance and tracking capacities that will enable ongoing evaluation and improvement. In addition, the health outcomes of children affected by these disorders should be monitored. Better understanding of response to clinical treatments and other interventions and the development of novel approaches to treatment are needed, particularly for rare disorders for which treatment protocols are less well defined. Surveillance programs can provide the research platform for both observational and experimental approaches to refine medical and other interventions for many of these disorders (8). The findings in this report are subject to at least three limitations. First, for the majority of the disorders screened, the cumulative incidence was derived from screening results for the approximately 2 million live births that occurred in the four states during 2001--2006. Although this number of births is sufficient to provide accurate estimates for many of the disorders (as evidenced by the relatively narrow 95% confidence intervals), the results observed among the four states might not be representative of the entire U.S. population (9). The analysis did account for some of this variability, particularly for hemoglobinopathies (which vary substantially by race and ethnicity) by using race- and ethnicity-specific rates to calculate the expected number of births. However, this approach was not possible as a general strategy because of lack of sufficient numbers of cases by race and ethnicity and lack of race- and ethnicity-specific information for Massachusetts. Second, an assessment of the accuracy of the rates for the rare disorders will not be possible until additional, population-based newborn screening data become available. Nevertheless, even if the estimated rates for the rare disorders were inaccurate by a factor of twofold or threefold, they would have only a modest impact on the estimated number of children with disorders identified using the expanded newborn screening panel. Finally, this analysis was not able to account for variations in the screening and diagnostic protocols among states that might have affected state-specific prevalences and estimates of the total number of cases. Newborn screening continues to be a critical public health program that ensures better health and developmental outcomes for newborns at high risk. The recent recommendation to expand newborn screening programs presents challenges in terms of 1) ensuring screening and follow-up for the many rare disorders and 2) facilitating the clinical care and management of complex and more common disorders (e.g., cystic fibrosis and hemoglobinopathies), which require different types of specialists and life-long clinical management. Acknowledgment The findings in this report are based, in part, on contributions by C Lagoy, National Center for Birth Defects and Developmental Disabilities, CDC. References
* Additional information available at http://www.marchofdimes.com/peristats/tlanding.aspx?dv=lt®=99&top=12&lev=0&slev=1. † Massachusetts, North Carolina, and Wisconsin. Data from California also were included because that state had conducted a rigorous pilot study (4) before it implemented expanded screening in 2005. § Additional information available at http://www2.uthscsa.edu/nnsis.
All MMWR HTML versions of articles are electronic conversions from typeset documents. This conversion might result in character translation or format errors in the HTML version. Users are referred to the electronic PDF version (http://www.cdc.gov/mmwr) and/or the original MMWR paper copy for printable versions of official text, figures, and tables. An original paper copy of this issue can be obtained from the Superintendent of Documents, U.S. Government Printing Office (GPO), Washington, DC 20402-9371; telephone: (202) 512-1800. Contact GPO for current prices. **Questions or messages regarding errors in formatting should be addressed to mmwrq@cdc.gov.Date last reviewed: 9/18/2008 |
|||||||||
|