 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
| ||||||||||
|
|
|
|
|
Persons using assistive technology might not be able to fully access information in this file. For assistance, please send e-mail to: mmwrq@cdc.gov. Type 508 Accommodation and the title of the report in the subject line of e-mail. Newborn Screening for Cystic FibrosisEvaluation of Benefits and Risks and Recommendations for State Newborn Screening ProgramsPrepared by Corresponding preparer: Scott D. Grosse, Ph.D., Senior Health Economist, Office of the Director, CDC/NCBDDD, 1600 Clifton Rd., NE, MS E-87, Atlanta, GA 30333; Telephone: 404-498-3074; Fax: 404-498-3070; E-mail: sgrosse@cdc.gov. Summary In November 2003, CDC and the Cystic Fibrosis Foundation cosponsored a workshop to review the benefits and risks associated with newborn screening for cystic fibrosis (CF). This report describes new research findings and outlines the recommendations of the workshop. The peer-reviewed evidence presented at the workshop supports the clinical utility of newborn screening for CF. Demonstrated long-term benefits from early nutritional treatment as a result of newborn screening for CF include improved growth and, in one study, cognitive development. Other benefits might include reduced hospitalizations and improved survival. Mixed evidence has been reported for pulmonary outcomes. Newborn screening in the United States is associated with diagnosis of CF a median of 1 year earlier than symptomatic detection, which might reduce the expense and anxiety associated with work-up for failure to thrive or other symptoms. Certain psychosocial risks for carrier children and their families (e.g., anxiety and misunderstanding) are associated with newborn screening. Exposure of young children to infectious agents through person-to-person transmission in clinical settings, although not an inherent risk of newborn screening, is a potential cause of harm from early detection. Involving specialists in CF care and infection control, genetic counseling, and communication can minimize these potential harms. Although screening decisions depend on a state's individual resources and priorities, on the basis of evidence of moderate benefits and low risk of harm, CDC believes that newborn screening for CF is justified. States should consider the magnitude of benefits and costs and the need to minimize risks through careful planning and implementation, including ongoing collection and evaluation of outcome data. IntroductionAfter sickle cell disease (SCD), cystic fibrosis (CF) is the second most common life-shortening, childhood-onset inherited disorder in the United States. Each year, approximately 1,000 persons in the United States receive a diagnosis of CF. Whether screening newborn infants for CF should be recommended on the basis of clinical benefit from early detection has long been the subject of debate and scientific investigation (1,2). In 1979, development of a test to measure immunoreactive trypsinogen (IRT) in dried blood spots, which is usually substantially elevated in newborns with CF, made universal newborn screening for CF feasible (3). Pilot screening for albumin in meconium had previously been carried out in certain European countries (4). The first newborn screening program for CF in the United States began in Colorado in 1982, followed by Wisconsin in 1985 and Wyoming in 1988. In 1997, CDC convened a workshop that reviewed the state of scientific evidence on newborn screening for CF and formulated recommendations (5). At that time, newborn screening for CF was conducted in those three states and in three other states (Connecticut, Montana, and Pennsylvania) in which certain hospitals provided screening for CF as a clinical service. Participants in the 1997 workshop found sufficient evidence of nutritional benefit to recommend state-based demonstration screening projects (5). Because evidence regarding pulmonary or other outcomes was limited, newborn screening for CF was recommended for research purposes, with informed consent and protocols for tracking and evaluating outcomes. Research was to focus on 1) the consequences of delayed diagnosis, 2) cognitive development caused by malnutrition, 3) pulmonary benefits, and 4) the cost-effectiveness of early detection through screening. A subsequent workshop was to be held to evaluate new evidence and, if warranted, to revise recommendations relating to newborn screening for CF. In November 2003, in cooperation with the Cystic Fibrosis Foundation (CFF), CDC held a second workshop in Atlanta, Georgia. This workshop had three objectives: 1) to review and evaluate the scientific evidence on benefits and risks of newborn screening for CF; 2) to review screening, diagnostics, and follow-up concerns in CF newborn screening decision making; and 3) to disseminate information about models and best practices for states that choose to adopt newborn screening for CF. The workshop was announced in the Federal Register, and the proceedings are available online (http://www.cdc.gov/ncbddd/cf/meeting.htm) (6). In addition, the majority of the papers presented at the workshop will be published in a CFF-sponsored supplement to the Journal of Pediatrics. Each state selects which disorders to include in its own newborn screening panel (7,8). As of August 2004, the number of disorders listed in state screening panels ranged from four to 40 (9). Criteria commonly used by states to decide on screening panels include the frequency of the disorder, the feasibility and accuracy of screening, the ability of early intervention to improve outcomes, the availability of treatment, and the cost of screening (10--12). Variability in screening criteria and how they are applied indicates a need for evidence-based assessments conducted at the national level (13). The traditional screening criteria have been used to justify universal newborn screening for disorders when early intervention is required soon after birth to prevent death or severe disability, as with phenylketonuria (PKU) (7). The application of these criteria to other disorders with less evidence of overwhelming immediate benefit poses challenges. Alternative frameworks for evaluating the balance of benefits and risks and approaches to screening that mitigate the risks might need to be considered for disorders such as CF (14). This report summarizes findings presented at the 2003 workshop and those of other studies in the peer-reviewed literature. It also presents recommendations to advise states that choose to adopt newborn screening for CF on how to maximize benefits and minimize harms in implementing screening. The information presented in this report is intended to help decision makers reach informed policy decisions. In addition, this report might be of use to federal agencies, professional organizations, industry, academic institutions, health-care providers, and the public. BackgroundGenetics and Pathophysiology of CFCF is an autosomal recessive genetic disorder; persons with CF have mutations in the gene encoding for the cystic fibrosis transmembrane conductance regulator (CFTR) protein on both alleles of chromosome 7. Although >1,000 mutations on the CFTR gene have been identified, a single common mutation, DF508, accounts for two thirds of all CF alleles worldwide (15,16). This mutation is particularly frequent in persons of northern European ancestry, who also have the highest rates of CF, and is less common among persons of other ancestries. Because different populations have different mutation frequencies, the sensitivity of a given DNA mutation panel for detecting persons with CF varies by race and ethnicity, and including mutations specific to racial and ethnic minority populations can improve detection of CF among those populations (16). Because of differing frequencies of CFTR mutations, the birth prevalence* of CF varies by race/ethnicity. On the basis of data from U.S. newborn screening programs, birth prevalence is 1/2,500--3,500 births among non-Hispanic whites, 1/4,000--10,000 births among Hispanics, and 1/15,000--20,000 births among non-Hispanic blacks (17--19). Non-Hispanic whites, who in 2000 accounted for 56% of births in the United States, constituted >90% of U.S. patients who received a diagnosis of CF (20,21). On the basis of data from state newborn screening programs that included CF in 2000, the overall birth prevalence of CF in the United States is approximately 1/3,700 (21). It is more common than PKU (1/20,000) and galactosemia (1/67,000) and less common than congenital hypothyroidism (1/2,500) and SCD (1/2,600)† (21). Mutations in the CFTR gene can alter the structure, function, or production of a cyclic adenosine-5'-monophosphate (AMP)--dependent transmembrane chloride channel protein that is critical for normal functioning of multiple organs. The organs and systems that are affected in CF include the lungs and upper respiratory tract, gastrointestinal tract, pancreas, liver, sweat glands, and genitourinary tract (15). For example, defective salt reabsorption in sweat glands leads to overly salty sweat and, in certain cases, to electrolyte imbalance, dehydration, and death. Deficient chloride transport in the lungs is thought to result in the production of abnormally thick mucus, which in turn is believed to lead to airway obstruction, neutrophil-dominated inflammation, and recurrent and progressive pulmonary infections (15). The combination of inflammation and infection accounts for the pulmonary symptoms related to CF (4). Acute viral respiratory infections, common to all children, are much more likely to develop into lower respiratory tract infections among children with CF, resulting in hospitalization and acquisition of chronic bacterial infections (22,23). Pancreatic insufficiency, which results from virtually absent pancreatic enzyme activity, is present at diagnosis among >80% of persons with CF and increases with age to >90%. Pancreatic insufficiency causes fat and protein malabsorption. Gastrointestinal symptoms associated with fat malabsorption include loose, foul-smelling fatty stools (steatorrhea) and abdominal pain. Nutritional consequences of pancreatic insufficiency include fat-soluble vitamin deficiencies and growth failure. Persons with CF who have normal or subnormal pancreatic enzyme activity, referred to as pancreatic sufficiency, rarely experience nutrition-related symptoms but are at risk for pancreatitis as they age (15). They also experience fewer pulmonary problems and have lower mortality (24,25). Research into genotype-phenotype associations in CF indicates that both pancreatic insufficiency and pancreatic sufficiency are associated with specific CFTR mutations. CFTR gene mutations have been placed into five classes. The first three are associated with complete loss of cyclic AMP-regulated chloride channel function and are identified as "severe" mutations. Mutations in the other two might allow for residual CFTR function and therefore are usually associated with milder phenotypes and pancreatic sufficiency (15,25). Persons who have two mutations from within classes I, II, or III almost invariably experience pancreatic insufficiency, and those with <2 mutations from classes IV or V usually maintain pancreatic insufficiency (25). The common DF508 mutation is a class II mutation that is associated with pancreatic insufficiency. Not all CFTR mutations have been identified or classified. Natural History and Diagnosis of CFThe first symptom of CF in approximately 15%--20% of children with CF is meconium ileus, an intestinal obstruction present at birth that usually requires surgery to correct (15). Children with meconium ileus have long-term adverse outcomes (e.g., malnutrition, lung disease, and mortality) at rates comparable to those for children detected later on the basis of clinical symptoms (26). Because meconium ileus is diagnostic of CF, screening does not increase early detection for these children. The most common symptoms of CF among children without meconium ileus include recurrent cough, wheezing, chronic abdominal pain, loose stools, and failure to thrive (15,27). Failure to thrive is particularly common in infants and young children. On the basis of data from the Wisconsin randomized controlled trial of newborn screening for CF during 1985--1994, the mean height-for-age Z-score at age 1 year among children without screening results is approximately --1.0, or 1 standard deviation (SD) below the reference mean (28). With the introduction of nutritional therapies (e.g., pancreatic enzyme supplements and a high-fat diet) after a diagnosis of CF with pancreatic insufficiency, the majority of children experience partial catch-up growth, and the mean height-for-age Z-score settles around --0.4 by age 2 years. Respiratory symptoms and infections increase with age. For example, for a cohort of children with CF followed in Wisconsin, the median age at which a pattern of frequent cough became present was age 10.5 months, and 75% had chronic cough by age 6 years (24). By age 7 years, 15%--25% of these children had lung function below the normal range, with progressive decreases in lung function as the children aged (26,29). Approximately 20% of children had positive cultures for Pseudomonas aeruginosa, the leading respiratory pathogen in CF, by age 1 year (30), but age at first acquisition varied widely. In the Wisconsin study, among subjects followed at the center that used recommended infection-control practices, the median age at first acquisition of P. aeruginosa was age 5 years (29). Infection with mucoid P. aeruginosa leads to irreversible pulmonary damage and death (31). In one cohort study, survival to age 16 years was 53% among children who had chronic P. aeruginosa infections and 84% among those who did not (32). Mortality in CF is associated with chronic obstructive pulmonary disease, with respiratory failure being the primary cause of death among >90% of persons with CF (15). Until the 1980s, deaths typically occurred during childhood or adolescence, but the majority of persons with CF now survive into adulthood. During 1974--1994, the international median age of death from CF increased from age 8 years to age 21 years (33). In the United States, the median age of death from CF in 2000 was age 24 years, with 5% of deaths occurring before age 10 years, 25% before age 17 years, and 75% before age 35 years (34). Because of increasing life expectancy, median age of death understates survival; during 1969--2001, the median predicted age of survival increased from age 14 years (35) to age 33 years (20). Early clinical recognition of CF on the basis of symptoms is desirable but difficult because the majority of symptoms are not specific to CF. Consequently, affected children often initially receive a diagnosis of food allergies, celiac disease, asthma, or bronchitis rather than CF. Misdiagnoses can entail multiple office visits, unnecessary diagnostic tests and hospitalizations, considerable cost to the health-care system, and anxiety for parents. This process has been referred to as a diagnostic odyssey, and narrative reports have documented resulting parental distrust of and anger toward health-care providers (36). The median age of diagnosis among all persons with CF in the United States is age 5.3 months (interquartile range [IQR]: 0.7--38.0 months) (27). The overall median age at diagnosis includes infants who receive a diagnosis of CF soon after birth on the basis of meconium ileus, newborn screening, prenatal screening, or a family history. The median age of clinical diagnosis on the basis of signs and symptoms other than meconium ileus is 14.5 months (IQR: 4.2--65.0 months), compared with 0.2 months (IQR: 0--0.9 months) for meconium ileus and 0.5 months (IQR: 0--0.9 months) for newborn screening (27). Diagnosis on the basis of symptoms among infants is associated with a >2-fold greater risk of medical complications before diagnosis than diagnosis resulting from screening (27). Delay between the appearance of CF-related symptoms and diagnosis can result in adverse psychosocial effects for the child's family (36). The traditional case definition for CF was based on a sweat chloride level of >60 mEq/L from pilocarpine iontophoresis (sweat test) and the presence of pulmonary disease or pancreatic insufficiency. Sweat testing consists of electrical-chemical stimulation of skin to produce sweat, collection of sweat on gauze pads or filter paper, and laboratory analysis of chloride content in collected sweat. In 1999, a CFF consensus panel developed a new case definition for CF based on multiple criteria: the presence of >1 characteristic phenotypic feature or a history of CF in a sibling or a positive newborn screening test, together with laboratory evidence of a CFTR abnormality as documented by 1) elevated sweat chloride concentrations, 2) identification of two CFTR mutations associated with CF, or 3) in vivo demonstration of characteristic abnormalities in ion transport across the nasal epithelium (37,15). Although a sweat chloride level of 60 mEq/L is diagnostic, infants with CF often have initial sweat values of 30--59 mEq/L (38,39). Sweat testing can be performed accurately on the majority of infants at age 2--3 weeks; however, not all infants have sufficient quantities of sweat for reliable testing (40). Classic CF, which usually manifests in early childhood, is associated with lung disease and pancreatic insufficiency. Certain persons with mild CFTR dysfunction, who often receive a diagnosis as adolescents or adults, are said to have nonclassic or atypical CF (15), defined as having sweat chloride results below the diagnostic level, milder phenotypic symptoms, and two identified CFTR mutations, at least one of which is associated with less severe disease (40). Sweat chloride levels in infants of 30--59 mEq/L are ambiguous, because persons with sweat chlorides in this range might have either classic or nonclassic CF, or they might not have received a diagnosis of CF (38,39). Because cohorts detected by screening might contain more persons with nonclassic CF with milder phenotypes, benefits of screening could be overstated. Ultimately, a better understanding of the natural history of the disorder in infants with two CFTR mutations and borderline or normal sweat test results is needed to provide appropriate education, counseling, treatment, and follow-up. Treatment of CFChildren with diagnosed CF receive different treatments, depending on the type of health-care facility and individual characteristics. Children who are identified as having pancreatic insufficiency receive pancreatic enzymes and fat-soluble vitamin supplements. Specialized CF centers vary in their approaches to the use of antibiotics for treatment and, in certain places, prophylaxis of respiratory infections (41,42). Persons with CF receive antibiotic treatment (oral, inhaled, or intravenous) for pulmonary exacerbations, characterized by increased cough and sputum production. Regular microbiologic monitoring and aggressive antibiotic treatment of infections have been demonstrated to reduce the number of children who experience chronic P. aeruginosa infections (43). Physiotherapy and inhalation of nebulized mucolytics are also common components of treatment regimens. To receive optimal treatment, children with CF should be examined at specialized CF centers that offer a comprehensive, multidisciplinary approach to CF care and that can closely monitor the development of respiratory infections and provide nutritional and psychosocial support (4). CFF accredits a national network of such centers, publishes clinical practice guidelines, and convenes consensus panels on CF care. Changes in CF care since the early 1980s have included 1) use of enteric-coated pancreatic enzymes to reduce the frequency of both gastrointestinal symptoms and nutritional complications, 2) replacement of low-fat diets with high-fat diets to offset the higher rates of fat malabsorption, 3) improved mucolytics and airway clearance techniques, and 4) use of more effective antibiotics to treat lung infections. These changes have contributed to substantial improvements in measures of malnutrition, lung function, and mortality among children and adolescents with CF. For example, during 1985--2001, predicted survival of persons with CF in the United States increased from age 25 years to age 33 years (20). During 1985--1999, mortality rates for children aged 2--15 years declined 45%--70%, mortality rates for persons aged 16--20 years were stable, and mortality rates for persons aged 21--40 years decreased 14%--20% (44). Recent pharmacologic advances hold the promise of more improvements in the health of persons with CF. For example, palivizumab, a monoclonal antibody against respiratory syncytial virus (RSV), has been demonstrated to be safe and effective in reducing RSV infections and hospitalizations among infants and young children at high risk (45). Hospitalization is one of the leading risk factors for the early acquisition of P. aeruginosa (23). Since 1994, new approaches have become available to slow the progression of pulmonary damage or to treat pulmonary bacterial infections in persons with CF (e.g., recombinant human DNase, inhaled tobramycin, and azithromycin) (46--49). Antibiotics and anti-inflammatory drugs have been reported to slow decline in lung function (31,50). Research is ongoing to determine the appropriate use of such drugs in infants and young children. In addition, two new therapies that directly target the mechanism of CF disease by improving chloride ion transport are in clinical trials (Preston Campbell, M.D., Cystic Fibrosis Foundation, personal communication, May 3, 2004). Prenatal Screening for CFCF can be identified before birth through prenatal screening and after birth through newborn screening. In 2001, the American College of Obstetricians and Gynecologists recommended that pregnant women, especially those in higher-prevalence populations defined by ethnicity, be offered screening for CFTR mutations (51). Carrier couples can choose prenatal diagnosis to determine whether a fetus has CF (defined by the presence of each parent's CF-associated mutation). Carrier testing for CF is offered by an unknown number of prenatal health-care providers. Preliminary data indicate that <20% of pregnant women in the United States receiving prenatal care are being screened for CF (52). Both the 1997 and 2003 workshops restricted their focus to newborn screening for CF as a state public health policy and did not address prenatal screening. The coexistence of two different screening programs for the same disorder deserves attention because of potential reciprocal effects and the opportunity for coordination of information provision. Prenatal screening for CF in other countries is reported to have led to a decrease in CF birth prevalence (53), but the majority of infants with CF will not be detected through prenatal screening. The sensitivity of prenatal screening for CF among the white population is <78% (54), much lower than that for newborn screening, and the sensitivity of prenatal testing in racial and ethnic minority populations is lower.§ Research is needed to evaluate the effect in this country of offering carrier testing in prenatal care on newborn screening and vice versa. Newborn Screening for CFAs of 1997, three states (Colorado, Wisconsin, and Wyoming) had universal newborn screening programs for CF, and in three other states, screening was conducted at hospital discretion by a state public health laboratory (Montana), academic laboratories (Connecticut), or a commercial laboratory (Pennsylvania) (55). During January 1998--April 2004, universal CF newborn screening was begun in four additional states (Massachusetts, Mississippi, New York, and New Jersey). Two more states (Oklahoma and South Carolina) are expected to start universal screening for CF during 2004 (Table 1). In 2000, approximately 400,000 (10%) children born in the United States were screened for CF; this number is expected to increase to 800,000 by the end of 2004 (12). Health-care providers can also order testing from private laboratories for disorders such as CF that are not included in state screening panels. Screening Protocols and Algorithms Multiple protocols and algorithms are used to screen newborns for CF (Table 1) (Box 1). All protocols begin with a first-tier phenotypic test that measures IRT in dried blood spots. Infants who have an elevated IRT measurement are then referred for further testing. The specific value used to decide whether IRT is sufficiently elevated to warrant further testing varies. Different lab kits for IRT produce varying distributions of IRT measures, and screening programs set cutoffs on the basis of evaluations of specimens from their own populations and the screening protocols and algorithms used (Table 2). Screening programs in five states (Colorado, Connecticut, Montana, New Jersey, and Wyoming) set absolute cutoffs for a normal IRT value on the first newborn blood spot (range: 90--105 ng/mL). Programs in four other states (Connecticut, Massachusetts, New York, and Wisconsin) classify as abnormal specimens in the top 4%--5% of IRT values for a given day or month, and programs in two states (Mississippi and Pennsylvania) use the top 10% of daily IRT values (55). Programs in five states (Colorado, Connecticut, Montana, South Carolina, and Wyoming) use an IRT-repeat IRT protocol. This protocol requires obtaining a second dried blood spot specimen at approximately age 2 weeks. At this age, elevated IRT values are more specific for CF because IRT values decrease with age in infants without CF. In Colorado, a routine second specimen is collected from all newborns, whereas in the other states, an elevated IRT on the first specimen necessitates collection of a second specimen from those infants. If the repeat IRT is elevated, with cutoffs varying among states, the child is referred for sweat testing. A sweat chloride of >60 mEq/L, confirmed by a second elevated sweat test result, is considered diagnostic for CF (37), although infants with CF might have lower sweat test results (38,56). In eight states (Connecticut, Massachusetts, Mississippi, New Jersey, New York, Oklahoma, Pennsylvania, and Wisconsin), screening programs use an IRT/DNA algorithm in which a single elevated IRT test is followed by DNA analysis on the same blood spot specimen to detect >1 specific CFTR mutation (55). Those newborns with a positive screening test (i.e., elevated IRT test and >1 detected mutation) are usually referred to a diagnostic center for a sweat chloride test to make a definitive diagnosis, although children with two disease-causing mutations are considered to have CF, regardless of the results of the sweat test (37). The program in one state (New Jersey) tests for only the common DF508 mutation although second specimens are requested for a repeat IRT test if no mutation is detected. Programs in three other states (Connecticut, Mississippi, and Pennsylvania) test first for the DF508 mutation and, if one DF508 mutant allele is detected, apply a multiple-mutation panel to screen for other mutations. Programs in the remaining four states (Massachusetts, New York, Oklahoma, and Wisconsin) use or plan to use a multiple-mutation panel to test all specimens with an elevated IRT. Programs in all seven states (Connecticut, Massachusetts, Mississippi, New Jersey, New York, Pennsylvania, and Wisconsin) that have implemented IRT-DNA algorithms also include a protocol that identifies infants without an identified mutation but with a highly elevated IRT value as being at risk for CF, because a child might have mutations for which a test is not administered. The cutoff of a highly elevated IRT is variable; cutoffs representing an IRT value of 99.8% or 99.9% of screened newborns or absolute values of 130, 170, or 200 ng/mL are used in different programs (55). Programs in three states (Massachusetts, New Jersey, and New York) treat such infants as positive screens and request that they be referred for sweat testing unless a repeat specimen with a normal IRT is first received. Programs in the remaining four states (Connecticut, Mississippi, Pennsylvania, and Wisconsin) report the results but do not request referrals for sweat testing (55). Screening programs that use mutation testing as part of their screening algorithm can choose from commercially available reagents for detection of multiple mutations (17,57), all of which have been developed to address the prenatal testing market's response to recommendations from the American College of Medical Genetics (ACMG) (58). The ACMG panel was chosen on the basis of the relative frequency of CFTR mutations among U.S. residents with CF (58) but did not take into account genotype-phenotype associations among persons with varying levels of severity of CF (25) or mutations detected with high frequency among certain racial/ethnic populations (59). Inclusion of a greater number of CFTR mutations in newborn screening panels will increase the number of persons identified as carriers and the numbers receiving either an ambiguous diagnosis of CF or a diagnosis of classic CF (17). Determination of which mutations should be included in panels for newborn screening should be developed by a workgroup of newborn screening specialists and persons with CF expertise. Analytic Validity The analytic validity of testing indicates the extent to which laboratory test results are reported accurately. Certain analytic errors might be detected through program follow-up; for example, during 21 years of screening, the Colorado program has reported four false-negative screens resulting from laboratory errors among 1,153,339 live births (18). To monitor analytical validity on an ongoing basis, newborn screening laboratories should participate in a proficiency testing program. Starting in 2002, CDC implemented a proficiency testing program for monitoring laboratory performance in testing IRT and DF508 mutations in newborn dried blood spots (60). A separate proficiency testing program for clinical laboratories performing CF carrier testing by using multiple mutation panels is also available (61). Clinical Validity The clinical validity of a screening test refers to the ability of a test to detect a person with a particular disorder. Determination of the clinical validity of a screening test requires the ascertainment of missed cases detected clinically as well as the tracking of final diagnostic status of all those with positive screens. Newborn screening algorithms for CF routinely result in false-negatives (62). Certain persons with CF either have IRT values below the cutoff used for screening (63) or are missed in the DNA stage of IRT/DNA screening because they do not have a mutation included in the mutation panel used by the screening program. Data on the clinical sensitivity of newborn screening for CF from screening programs in the United States have been published (Table 2). Children with meconium ileus might have low IRT values and be missed by screening but are detected soon after birth anyway (4); 90%--95% of children with CF without meconium ileus are reported to be detected by IRT-repeat IRT screening (64--66,18). IRT/DNA screening and follow-up protocols that use multiple mutation panels and directly refer children with extremely high IRT values for sweat testing have achieved clinical sensitivity of >98% (17). The number of false-positive¶ results in newborn screening tests for conditions other than CF is often >50 times the number of cases of diagnosed disease (64). In comparison, the number of false-positive results from newborn screening for CF is moderate. Among states using IRT/DNA algorithms, the ratio of false-positive screens to true-positive cases is reported to be 9.5:1 in Wisconsin (57), 12:1 in Massachusetts (67), and 25:1 in New York (Kenneth A. Pass, Ph.D., New York State Department of Health, personal communication, August 19, 2004) (Table 2). In Colorado, which performs repeat IRT measures on two specimens, five false-positive CF screening results are reported for every true-positive, whereas if positive results had been reported based on an elevated IRT value from the initial specimen, the average ratio of false-positives to true-positives would have been 30:1 (18). Although the number of false-positive screens for CF is reasonable relative to other newborn screening tests, screening for CF carries an additional responsibility for providing genetic counseling to families of screen-positive children who actually carry a CFTR mutation despite having a negative diagnostic test. Reporting of CF carrier status is one outcome of newborn screening, which has the potential to be both a benefit and a risk for families of carriers. Because newborn screening identifies considerably more carriers than children with CF, benefits and harms to families of carriers should be considered in assessing newborn screening for CF. Costs The laboratory cost of CF newborn screening varies with the screening algorithm but is comparable to that of newborn screening tests in common use (64). Data from the Wisconsin screening program indicate that the laboratory cost of IRT screening is $1.50/test, the cost of a single-mutation analysis is $20.50, and the cost of a multiple-mutation test is $50.70 (68). These numbers yield average costs of $2.35 for an IRT/DNA algorithm with a single mutation and $3.60 for an IRT/DNA algorithm with a multiple-mutation panel. A full accounting of the costs of implementing newborn screening for CF would require additional information on the costs of follow-up, diagnosis, counseling, and providing care. In addition, averted diagnostic and treatment costs, if any, should be factored in. A preliminary analysis indicates that averted diagnostic costs could cover the majority of initial screening costs (69). Scientific EvidenceSelection of StudiesIn preparation for the workshop, CDC searched the MEDLINE® database of medical literature for articles on newborn screening for CF published since the 1997 workshop that discussed health outcomes or psychosocial outcomes in groups of children identified with CF through newborn screening in comparison with children identified through other means. In addition, review articles were obtained and reference lists searched to identify additional articles. Subsequently, a decision was made to include findings from previously published studies, which were identified by searches of reference lists. An analytic framework was developed that modeled indirect links from newborn screening to nutritional status and from nutritional status to lung function and survival (Figure). Accordingly, a literature search was also conducted to locate studies of associations between nutritional status among children and adolescents with CF and lung function or survival outcomes. This search used the MEDLINE® database and soliciting of specialists for additional studies. A planning committee for the workshop identified researchers worldwide examining outcomes in relation to newborn screening for CF. At least one person from each research study group was invited to present the study's findings at the 2003 workshop. Certain presentations were based on research that had already been accepted for publication or was in press. In addition, CFF decided to cosponsor a peer-reviewed supplement to the Journal of Pediatrics to which presenters were invited to submit papers for publication; 15 papers cited in this report are scheduled for publication in that supplement. Analytic Framework for Evaluating Effects of CF Newborn ScreeningTo evaluate the potential benefits and risks of newborn screening for CF, CDC applied an analytic framework to interpret evidence of clinical utility (i.e., the net balance of health outcomes) of earlier identification and treatment (Figure). This framework draws in part on the approach used by the U.S. Preventive Services Task Force (USPSTF) (70). It considers both potential benefits and harms from screening; harms are classified separately as adverse effects from screening and those from diagnosis or treatment. The benefits of screening flow from early, asymptomatic detection and can be classified in terms of health benefits to the affected person and psychosocial benefits to persons and families. To classify health benefits, CDC used the Strength of Recommendations Taxonomy (SORT), a recently proposed patient-centered approach to grading evidence in medical literature (Box 2) (71). The SORT framework does not include psychosocial outcomes, which constitute key benefits and risks from newborn screening for CF and should be considered in policy recommendations. The potential psychosocial risks of screening include factors associated with 1) false-positives (e.g., unnecessary testing and possibly unnecessary treatment for the child, undue parental anxiety, and desensitization of providers), 2) false-negatives (e.g., potential delay in diagnosis for child and false reassurance for patients), 3) carrier reporting (e.g., possibly unwanted information and fear of stigmatization or insurance discrimination), and 4) misinformation (e.g., errors in communication or misunderstanding of results). Potential harms to CF patients of early detection and treatment as a result of newborn screening include side effects of therapies (e.g., drug resistance and toxicities) and earlier exposure (through person-to-person transmission from older children with CF) to bacteria associated with chronic airway infection in CF. The SORT taxonomy is used to assess the clinical effectiveness of interventions based on a structured review of research findings (71). The SORT framework categorizes studies into three levels (Levels 1, 2, and 3) on the basis of study design and type of outcomes assessed. The total evidence for an intervention is given one of three grades (A, B, or C) on the basis of the assigned levels of the individual studies. The SORT taxonomy distinguishes two classes of health outcomes: 1) disease-oriented outcomes (e.g., intermediate, histopathologic, physiologic, or surrogate results) that might reflect improvements in patient outcomes and 2) patient-oriented outcomes (e.g., reduced morbidity, reduced mortality, symptom improvement, improved quality of life, or lower cost) that help patients live longer or better lives (71). In the SORT framework, either a high-quality randomized controlled trial (RCT) or a meta-analysis of RCTs that demonstrates improved patient-oriented outcomes is considered Level-1 evidence. Information on patient-oriented outcomes from a lower-quality clinical trial, cohort study, or case-control study constitutes Level-2 evidence. All other types of research studies, including case series, are classified as Level 3, along with all studies, even RCTs, that provide information restricted to disease-oriented outcomes. For the SORT framework to be applied, endpoints used in evaluations of newborn screening for CF are classified as either patient-oriented or disease-oriented. The approach taken in this report is to classify endpoints that are collected during routine clinical monitoring of individuals with CF as disease-oriented outcomes. These include measures of nutritional and pulmonary outcomes (e.g., height and weight, spirometric measures of lung function, and chest radiograph scores). In this analysis, survival, quality of life, and cost (including hospitalizations and invasive therapies) were classified as patient-oriented outcomes. Cognitive function, which is not routinely assessed in persons with CF, was also classified as a patient-oriented outcome because of its direct link to quality of life and because it is not a surrogate outcome in CF. The classification of certain endpoints as disease-oriented or patient-oriented outcomes has implications for assessment of evidence on newborn screening for CF. In particular, growth retardation might be regarded as both a patient-oriented outcome and a disease-oriented outcome. The high demand for expensive growth hormone therapy, which results in moderate gains in linear growth for children with CF who have low height-for-age (72,73), indicates that below-normal stature might be viewed as a patient-oriented outcome. In addition, growth retardation among children with CF has been demonstrated to be a strong predictor of survival. An RCT indicating reduction in growth retardation would be classified as Level-1 evidence if this outcome were classified as patient-oriented but as Level-3 evidence if it were classified as disease-oriented. The SORT approach integrates the strength of evidence approach (74) with the type of health outcomes considered in establishing a hierarchy of evidence (71). In the SORT taxonomy, an A-level recommendation requires consistent and high-quality, patient-oriented evidence, including consistent findings from at least two high-quality randomized controlled trials. A B-level recommendation requires patient-oriented evidence (i.e., an improvement in morbidity, mortality, symptoms, quality of life, or cost) based on Level-1 or Level-2 evidence. A C-level recommendation is based on evidence relating to disease-oriented outcomes from any type of study or for patient-oriented outcomes from other types of observational studies, case series, or opinions of specialists (71). A limitation of the SORT framework for evaluating the overall strength of evidence for newborn screening tests is the scarcity of RCTs. CF is the only condition for which two RCTs of newborn screening have been conducted. This puts newborn screening at a disadvantage, with a B-level recommendation realistically the highest that can be assigned to any newborn screening test by using this framework. This report summarizes what is known about the strength of evidence for health benefits of newborn screening for CF, including assessments of the strengths and limitations of study designs and the consistency and magnitude of benefits reported. However, the report is not a formal systematic evidence review that would form the basis for an evidence-based practice guideline (70,71,74,75). Systematic reviews involve a lengthy process in which teams of reviewers conduct structured reviews with blinded assessments of study quality. The only newborn screening tests endorsed by USPSTF on the basis of systematic reviews are for PKU, congenital hypothyroidism, and hemoglobinopathies (75). Evidence for BenefitsSources of DataInformation to assess the effect of newborn screening for CF on health benefits for children with CF is available from three types of investigations: 1) RCTs of newborn screening, 2) geographic and temporal comparisons of screened and unscreened cohorts, and 3) assessments of outcomes of persons with CF reported to registry databases. The methods of two randomized trials of newborn screening for CF and four observational studies comparing health outcomes among screened and unscreened cohorts are summarized (Table 3). RCTs The Wisconsin CF Neonatal Screening Project randomly assigned neonates born in Wisconsin during 1985--1994 to either a screened or control group (28,76) (Table 3). Neonatal CF screening was performed on blood spots from all subjects, but positive results were reported only to families in the screened group. Positive results were released to families in the control group if any of the following occurred: 1) parents requested the results, 2) CF was diagnosed conventionally, or 3) the child reached age 4 years and a diagnosis of CF had not been established. Subjects with a diagnosis of CF in both groups were entered (after informed consent) into a longitudinal protocol that specified evaluation methods and standardized therapeutic interventions. Caregivers and investigators were blinded to group identity after the first visit. The study protocol included follow-up after diagnosis with CF every 6 weeks during the first year of life and every 3 months thereafter, through age 17 years. All children received care at one of two CFF-accredited care centers. The Wisconsin RCT had two key limitations, both of which could lead to underestimation of the magnitude of health benefits of screening in that study. First, despite randomization of study subjects, substantially more subjects with DF508 alleles (p<0.05) or pancreatic insufficiency (p<0.05) were identified in the screened group (77). Second, during the first 6 years of the study, premature acquisition of P. aeruginosa occurred among screened children who were followed at one of the two CF care centers participating in the study that had a small, crowded waiting room in which infants and older children with established infections mixed (30). The median pseudomonas-free period was 52 weeks among screened infants seen at the first center and 289 weeks among those followed up at the second center, which segregated children enrolled in the study from other patients (76). In the United Kingdom, all neonates born in Wales and the West Midlands during 1985--1989 were randomly allocated to undergo or not undergo CF screening on an alternate-week basis (78) (Table 3). Because no screening was performed in the control group, an unknown number of undiagnosed cases of CF in this group were not ascertained. A review of registry data revealed previously unknown CF-related deaths among children in the unscreened group (79). No specific treatment and follow-up protocol existed in the study. Children who received a diagnosis of CF through screening were supposed to be followed annually at a CF care center until age 5 years; however, a substantial number of unscreened children with CF did not receive care at a CF care center (79). Underascertainment in the control group, lack of standardized treatment protocols, and short, uneven follow-up indicate that this is a lower-quality clinical trial that would yield Level-2 rather than Level-1 evidence. Cohort Studies An observational study in Australia compared outcomes among children with CF born in New South Wales during the 3 years before the introduction of CF newborn screening in July 1981 (and who therefore received a diagnosis conventionally) with those born during July 1981--July 1984 (80--82) (Table 3). All analyses were conducted on an intent-to-treat basis, with children included in the screened cohort if they were born while screening was offered, regardless of mode of diagnosis. All subjects were followed at a single CF clinic. Clinical data were collected retrospectively from medical records at diagnosis and ages 1, 5, 10, and 15 years. Changes in clinical practice over the 16 years of the study might have influenced comparisons. Improved nutritional practices introduced during 1981--1983 could potentially have biased outcomes in favor of screening (81). A pilot study used the albumin content of meconium to screen for CF in approximately half (45%) of all newborns born during 1973--1979 in a defined area in the northern Netherlands (83). Screening was not random; providers decided whether to offer screening, and parents could decide whether to participate. Investigators compared outcomes of the screened cohort (S) with two cohorts of children who received diagnoses clinically: one that received a diagnosis at the same time as the screened group (non-S) and one that received a diagnosis in the 6 years after the pilot screening project was discontinued (post-S) (83--85). CF care was provided at a local hospital with (54%) or without (46%) semiannual to annual visits at a specialized CF center. Follow-up and therapeutic protocols were not standardized. Clinical data were collected prospectively for 1980--1990 and retrospectively for 1990--1997, after an intent-to-treat analysis (85,86). The post-S group is not subject to selection bias, which might characterize the non-S group. More confidence can be placed in findings consistent for both comparison groups. An observational study from France compared children with CF born during 1989--1998 in Brittany, which screened newborns for CF, with a comparison group of newborns in a neighboring region, Loire-Atlantique, which did not implement screening for CF and had comparable CF care (87) (Table 3). Standardized follow-up was provided for patients who received a diagnosis of CF, including monthly clinic visits for the first 6 months after diagnosis and quarterly clinic visits thereafter. Therapeutic management was standardized. Differential underascertainment did not appear to be a major problem in this study, because the same birth prevalence was observed in both areas. Primarily because of differing population size, the S cohort (n = 77) was larger than the non-S cohort (n = 36). The intent-to-treat principle was not followed because false-negative screening results (n = 5) were excluded from the S cohort; however, the limited number of excluded patients is unlikely to have affected the conclusions. In addition, the exclusion of children with meconium ileus in Brittany (n = 14) and Loire-Atlantique (n = 10) affected the two cohorts disproportionately. Data from northeastern Italy have been used to compare outcomes among persons with CF detected by screening and symptoms. One analysis compared outcomes of persons with CF born during 1973--1981 in Veneto and Trentino and detected by newborn screening by meconium albumin testing with persons with CF diagnosed conventionally in Veneto and Trentino and two neighboring regions without screening (88) (Table 3). A second analysis compared outcome data on children with CF born during 1983--1992 in Veneto, where screening with CF was universal, with data on children with CF born in Sicily, where CF newborn screening was not practiced (88) (Table 3). Although the investigators reported that the two groups had similar frequencies of severe CFTR mutations and comparable treatment protocols, the two regions differ in socioeconomic status (89). Another analysis by other investigators compared persons with CF born during 1973--1992 who were examined at the CF center in Verona, distinguishing between those who received a diagnosis by newborn screening and those who received a diagnosis clinically (90). Cohort studies have at least two limitations that decrease their usefulness as evidence for a causal effect of newborn screening for CF. First, historic controls, as used in both the Australian and Dutch studies, are subject to bias because of improvements in CF treatment over time (89). In the case of the Australian study, such changes might overstate the apparent benefits of screening, whereas in the Dutch analysis, the effect would be in the opposite direction. Second, concurrent controls from different regions are subject to bias from variation in the quality of CF care provided across regions (89). Registry Studies Population-based CF registries in the United States and the United Kingdom have been used to model the effect of newborn screening on outcomes. The United Kingdom Cystic Fibrosis Database (UKCFD) was established in 1995 and includes data from 41 large specialist CF centers and 12 smaller CF clinics (91). One analysis of the UKCFD data compared outcomes for two groups of children aged 1--9 years, excluding those with meconium ileus: children who received a diagnosis on the basis of newborn screening before age 3 months (n = 184) and children who received a diagnosis on the basis of symptoms, excluding those with a positive family history (n = 950) (91). In the United States, the CFF National Patient Registry (CFNPR) contains information on >23,000 persons with CF cared for through a national network of CFF-accredited CF care centers and affiliated satellite clinics. These centers and their affiliates provide integrated, multidisciplinary care and have collaborated to develop standardized data-collection protocols (34). CFNPR data are made available by CFF to researchers and have been used in numerous statistical studies of outcomes. Registry data have at least three limitations. First, how persons were detected might be misclassified. For example, among 27,703 persons who appeared in CFNPR >1 time during 1986--2000, a total of 69 (8%) of 898 persons who reportedly received a diagnosis on the basis of newborn or prenatal screening did not receive their diagnosis until age >12 months (92), and 90 (10%) received a diagnosis before 1980 (93) even though screening was first introduced in the United States in 1982. Second, because persons detected by screening are more likely to have been born more recently and CF outcomes have improved over time, comparison of screened and unscreened groups that do not control for year of birth are subject to bias. Differences in race/ethnicity, genotype, pancreatic insufficiency, and birth year can be controlled in statistical analysis. Also, because newborn screening is closely correlated with region of birth, regional differences in quality of care might be difficult to separate from screening as an influence on CF outcomes. Finally, registry data, like all observational data, are subject to ascertainment bias (89). This is particularly a problem for studies that set a low cutoff for age of diagnosis (e.g., age 36 months). In the absence of screening, age at diagnosis varies substantially, and a person with milder clinical symptoms is more likely to receive a diagnosis at a later age. Disease-Oriented OutcomesNutritional Outcomes The most convincing evidence of health outcomes from screening for CF is in the area of nutrition and growth (19,89,94). In the absence of early treatment, infants and children with CF are subject to growth failure and prolonged vitamin deficiency. Anthropometric measures of nutritional status include height and weight, weight relative to height, and, for infants and young children, head circumference. The majority of studies have compared heights and weights to reference growth curves by using Z-scores, with a 1.0 difference in Z-score roughly corresponding to a difference of 1 SD in the reference curve (95). The absolute difference in weight or height for a given Z-score varies by age. In 1997, the Wisconsin Cystic Fibrosis Neonatal Screening Project demonstrated that diagnosis resulting from newborn screening and early intervention was associated with better growth (28). A subsequent analysis that included growth measurements before age 4 years for children in the control group unblinded at that age and with more years of follow-up provided conclusive findings (77) (Table 4). When pooled across all ages with a statistical model that controlled for covariates and interaction terms, children in the screened cohort were substantially taller (p<0.01) and slightly heavier (p = 0.06) than children in the standard diagnosis (control) cohort (62). Visual inspection of the graphs reveals that differences in mean height-for-age persisted through age 10 years but that the difference in weight-for-age disappeared after age 4 years (62). No differences in weight-for-height were observed at any age. The magnitude of the difference in height-for-age Z-scores varied among children aged 4--10 years (range: 0.2--0.5) (62). The U.K. RCT did not report substantial differences in either weight or height through age 4 years; however, <20 children were followed for 4 years (78). The difference in findings might also reflect differences in follow-up care, which was not standardized in the U.K. study. Decreased risk of chronic malnutrition or growth retardation, typically defined as being below the 5th or 10th percentiles for height or length relative to age, might be more important from a clinical perspective than mean height-for-age, because it is associated with increased risk of childhood morbidity and death (96--98). The percentages of children in the Wisconsin RCT being below the 10th percentiles for height-for-age and weight-for-age were substantially lower in the screened group, adjusting for covariates (e.g., age and genotype) (p<0.01) (77). In the control group, the percentage with low height-for-age declined from 40% at age 1 year to approximately 20% by age 4 years and remained stable, whereas the percentage in the screened group with low height-for-age was approximately 10%--15% through age 8 years. Among infants and young children, growth retardation without a known cause is referred to as failure to thrive. Failure to thrive is one of the leading developmental disorders among young children (97), as well as a common cause of pediatric hospitalization (98), and prevention of failure to thrive can reduce health-care costs and avert parental anxiety. As was true of the Wisconsin RCT, observational studies have reported greater height-for-age and, to a lesser extent, weight-for-age among children detected through screening. In Australia, repeated analysis of cross-sectional differences indicated that mean height-for-age Z-score was consistently greater by 0.4 among a cohort of 60 children born after screening was started in 1981 compared with a cohort of 57 children born during the 3 previous years. However, as a result of attrition, the differences were statistically significant only at age 1 year and age 5 years (p<0.05) (81).** At age 15 years, the mean difference in height was 3.4 cm (95% confidence interval [CI] = --0.4--7.2) and the mean difference in height-for-age Z-scores was 0.4 (95% CI = --0.1--0.9) (82). Data from the Dutch screening pilot study indicated a similar differential in height-for-age, with a difference in predicted height through age 12 among 18 children with CF in the screened group of 0.42 SD (95% CI = --0.20--1.04) compared with 24 unscreened children born during the same period; owing to the limited number of observations, this difference was not statistically significant (85). Compared with children born after screening, heights and weights were slightly but not substantially lower in the screened group, which could reflect better nutritional therapies available to the more recent birth cohorts (85). In France, children born in Brittany (where newborn screening has been in practice since the 1980s) had higher average height and weight Z-scores than those born in neighboring Loire-Atlantique (where screening was not in practice). Differences in height-for-age Z-scores, which were primarily 0.3--0.6 at each age, were statistically significant at ages 1, 3, and 5 years, and differences in weight-for-age were statistically significant at ages 1 and 8 years (p<0.05) (87). An analysis of UKCFD cross-sectional data for 2002 determined an overall difference in median height-for-age Z-score of 0.32 among U.K. children with CF detected through newborn screening compared with those detected on the basis of clinical symptoms (p<0.005) (91). By 3-year age groups, differences in height Z-scores were 0.39 at ages 1--3 years and 0.32 at ages 4--6 years (p<0.05), whereas a Z-score difference of 0.27 among those aged 7--9 years was not statistically significant. For the subset of children homozygous for the DF508 mutation, the difference in height-for-age Z-score was smaller (0.22; 95% CI = --0.01--0.44) and did not reach statistical significance. The difference in median weight-for-age Z-scores was smaller and not statistically significant in either analysis. In Italy, two analyses determined evidence of nutritional advantage from screening. First, a study conducted among children born in northeastern Italy during 1973--1981, when a pilot screening of meconium albumin was in place, revealed that those detected by screening, all with pancreatic insufficiency, had weight Z-scores better than the non--meconium ileus symptomatic group with pancreatic insufficiency at ages 4--6 years through ages 16--18 years (p<0.05); height Z-scores were significantly different from ages 6--8 years through ages 14--16 years (range: 0.6--1.0; p<0.05) (88). A second analysis compared all children born in Veneto during 1983--1992, when IRT screening was in place, with those born in Sicily, which had an intensive program of promoting early diagnosis on the basis of symptoms. Differences in height Z-scores were statistically significant at each 2-year age group from ages 0--2 years through ages 14--16 years (range: 0.4--0.8; p<0.01, except for ages 2--4 years) (88). Another nutritional benefit of newborn screening is earlier initiation of vitamin supplements. Deficiencies of fat-soluble vitamins (vitamins A, E, and K) are common among children with CF not receiving nutritional supplements (99); for example, half of infants in the Wisconsin RCT had vitamin E deficiency at diagnosis (77,100). Prolonged vitamin E deficiency can in certain cases lead to hemolytic anemia and other adverse outcomes, which can potentially be avoided through early diagnosis and intervention. These deficiencies can be corrected by nutritional therapy, and few children in either group in the Wisconsin trial had vitamin deficiencies when assessed later (77). An analysis of cognitive scores supported the hypothesis of a developmental effect of prolonged vitamin E deficiency among children with CF (100). Pulmonary Outcomes Existing measures that can be used to quantify early lung disease in young children with CF are limited. Lung function, assessed by spirometry, is the most common surrogate measure of obstructive lung disease and is predictive of survival in adolescents and adults with CF (101--103). The most frequently used measure of lung function in CF is forced expiratory volume in 1 second (FEV1) as a percentage of a predicted value based on height and age, which is usually measured only among children aged >5 years because of the limited ability of younger children to cooperate reliably with the testing. Other pulmonary outcome measures used among children with CF that might be indicative of the extent of lung disease are chest radiograph scores (e.g., Brasfield, Wisconsin, and Northern), which measure structural damage to the lungs, and Shwachman clinical scores of overall disease severity. Both types of measures involve subjective assessments. The Wisconsin study data indicate that chest radiograph scores might be a sensitive measure in young children, with half of children demonstrating signs of potentially irreversible lung damage by age 2 years (24). Although high-resolution chest tomography is an even more sensitive measure of early CF lung disease, this has not been used as an outcome measure in newborn screening evaluation studies. Lung function. Using spirometric measures, the Wisconsin RCT identified no statistically significant difference in lung function between patients in the screened and standard diagnosis groups through age 16 years (29). At age 7 years, 88% in the screened group and 75% in the control group were in the normal range (>89% predicted FEV1), and relatively limited deterioration with age was noted in either group. The U.K. RCT did not follow children long enough to collect spirometric measures of lung function. With one exception, no observational studies have identified statistically significant differences in lung function associated with newborn screening. An Australian study identified significantly better lung function among patients in the screened group, including assessments performed at ages 5 (mean difference in predicted FEV1 score: 9.0%; p<0.01), 10 (mean difference in predicted FEV1 score: 9.4%; p<0.05) (81), and 15 (mean difference in predicted FEV1 score: 12.3%; p<0.01) years (82). The differential in lung function was greatest at age 15 years, a mean difference in predicted FEV1 scores of 12.3% (95% CI = 2.9--21.7). In the Dutch study, the differential in FEV1 between the screened group and those who received a diagnosis conventionally during the same period (1973--1979) was similar in magnitude, 8.6 (95% CI = --6.0--23.2) but was not statistically significant. Virtually no difference was observed in lung function between the screened group and those who received a diagnosis conventionally in the 6 years after the newborn screening pilot project ended (85). Similarly, in both the French study and the UKCFD analysis, no differences in lung function were identified between S and non-S cohorts (87,91). Analyses of CFNPR data likewise have found no clear evidence of a benefit of newborn screening or early asymptomatic detection in terms of lung function (26,93).†† Chest radiograph and clinical scores. Investigators for the Wisconsin RCT longitudinally analyzed chest radiograph scores by using two raters for each chest film and two different scoring methods to calculate reliable measures of progression of lung disease (24,29). An analysis indicated that differences in chest radiograph scores were, if anything, to the disadvantage of the screened group, except at time of diagnosis. Chest radiograph scores measured at the same ages were similar between the two groups before age 10 years but were substantially worse in patients in the screened group at age >12 years (P<0.05) (29). The greater rate of decline in chest radiograph scores among screened patients occurred only among those positive for P. aeruginosa. This probably was attributable to a significantly earlier colonization with P. aeruginosa infection (52 versus 289 weeks) in the screened group followed at the clinic that in the early years of the study integrated young and old patients irrespective of their pulmonary status (29). The U.K. RCT did not find differences in clinical or radiological scores between groups at ages 1--4 years (78). Observational data on lung disease based on subjective radiographic or clinical assessments appear more supportive of an apparent pulmonary benefit of newborn screening for CF, but subjective, unblinded assessments could be influenced by knowledge of the screening status of the child. Three studies have reported better radiographic outcomes for screened children (Table 4). The Australian study reported significantly better Shwachman chest radiograph scores at age 15 years among patients in the screened group (2.3; 95% CI = 0.3--4.2), although not at ages 5 or 10 years; the difference in Shwachman clinical scores was statistically significant at ages 5 (3.8; p<0.01), 10 (5.3; p<0.05), and 12 (7.0; p<0.05) years (81,82). In France, both Brasfield chest radiograph scores and Shwachman clinical scores were significantly better among screened children (p<0.05) at each age; Brasfield scores among screened children declined 8% as children aged 1--9 years, compared with a 20% decline among unscreened children (87). Lastly, an analysis of UKCFD data has reported significantly better Northern chest radiograph scores (1.0; 95% CI = 0.0--0.99) and Shwachman clinical scores (3.0, 95% CI = 1.0--4.0) in neonatally screened patients ages 1--9 years (p<0.05) (91). Significant differences of comparable magnitude were apparent when the analysis was restricted to DF508 homozygotes to control for potential confounding by genotypic differences. Respiratory microbiology. In principle, newborn screening can allow for interventions to delay the onset of chronic P. aeruginosa colonization. A study from Denmark has reported that frequent culturing of specimens from persons with CF and aggressive treatment of first infections with P. aeruginosa avoids or delays the onset of chronic colonization (31). An analysis of UKCFD data has reported lower rates of both transient and chronic P. aeruginosa infection among young children detected by newborn screening (91). Among children aged 1--3 years, only half as many children in the screened group had at least one positive P. aeruginosa culture, both overall and among DF508 homozygotes (p<0.01); among DF508 homozygotes, a significantly lower proportion of children had three or more positive cultures (p<0.05) (91). In the United Kingdom, at least one CF center that participates in newborn screening also provides aggressive interventions against P. aeruginosa infections (43); the statistical finding could possibly reflect an association of newborn screening with variations in CF care. However, among children aged >3 years, which would include children with milder CF diagnosed symptomatically at later ages, no difference in rates of positive cultures was observed (91). Studies from France and the United States have not reported lower rates of isolation of P. aeruginosa from respiratory cultures taken from screened children with CF, including the Wisconsin RCT (76,87). An analysis of CFNPR data reported that although children with early asymptomatic detection had a lower frequency of colonization with this pathogen overall, after state of birth and other variables were controlled for, these children did not have a lower risk (104). Similarly, an analysis of CFNPR data reported that risk of earlier acquisition of P. aeruginosa among children detected symptomatically did not differ significantly from those detected by screening after controlling for confounding variables (e.g., genotype) (93). An analysis of CFNPR data from 2000--2002 determined that 15% of infants with CF detected by newborn screening had positive cultures for P. aeruginosa, compared with 25%--29% among infants detected on the basis of symptoms, but fewer than half of persons with CF detected by symptoms received a diagnosis as infants (27). Patient-Oriented OutcomesSurvival Because of low CF mortality rates in childhood, a substantial number of subjects would be required to detect statistically significant differences in survival between screened and unscreened cohorts (5). Also, including deaths unrelated to CF and deaths among children with meconium ileus might mask benefits in terms of preventable CF-related deaths. Data from the the U.K. RCT supplemented by ascertainment of CF-releated deaths from the UKCFD and other sources indicate that significantly fewer CF-related deaths occurred among the screened cohort (79); four CF-related deaths were recorded before age 5 years among 71 children who were selected randomly not to be screened, and no early CF-related deaths were reported among 78 children selected randomly to be screened (p<0.05). However, two of the four deaths occurred among children who received a diagnosis by age 7 weeks and might not have been preventable by screening. The other two deaths occurred among children who did not receive a diagnosis before death, which highlights prevention of undiagnosed CF mortality as a potential benefit of screening (86). Whether undiagnosed CF mortality is widespread in the United States is not known. Wisconsin study investigators examined death records for children who were born during the study period (62) but determined that no children had died with CF before receiving a diagnosis (Philip M. Farrell, M.D., Ph.D., University of Wisconsin Medical School, Madison, personal communication, November 20, 2003). No survival data from the Wisconsin RCT have been published. Observational studies have consistently identified lower mortality rates in screened relative to unscreened cohorts. Studies in Australia (82) and the Netherlands (85) used an intent-to-treat approach. In the Australian study, CF-related mortality among children aged <15 years was 47% lower among the screened cohort (6.7%) than among those born before screening (12.7%); this difference is not statistically significant, although deaths among the screened cohort occurred an average of 4 years later (p<0.05) (82). In the Dutch study, the relative risk for dying through age 12--24 years for 24 persons in the screened group was 0.43 (95% CI = 0.13--1.38) compared with 29 persons in the unscreened group born during the same period and 0.35 (95% CI = 0.09--1.41) compared with 34 persons with CF born after screening was terminated (85). Even greater relative differences in survival within 6--12 years after birth were apparent, but no separate statistical analysis was reported. Two observational studies from Europe have reported statistically significant differences in CF mortality comparing regions with and without newborn screening for CF. An analysis of Italian data that compared mortality among 126 children with CF born during 1983--1992 in the northeast in Veneto, which screened for CF, with mortality among 152 children born in the south in Sicily, which did not screen for CF, indicated that two deaths occurred among the former population and 18 among the latter (88). However, two analyses of data from Veneto and neighboring regions in Italy did not find mortality before age 10 years to be substantially lower among children with CF detected by screening (88,90). A similar analysis in France did not follow an intent-to-treat analysis but compared children detected by screening without meconium ileus born in two regions with those who received a diagnosis on the basis of symptoms other than meconium ileus. Three CF-related deaths occurred among 36 children born in Loire-Atlantique during 1989--1998 who were detected on the basis of symptoms other than meconium ileus, and none occurred among 77 children without meconium ileus in neighboring Brittany who were detected through screening, a statistically significant difference (p<0.05) (87).§§ Both studies reported similar prevalence of CF and similar CF treatment protocols across regions (87,88). In the United States, analysis of CFNPR data during 1986--2000 identified a higher risk for death among persons with CF detected by symptoms rather than by screening (p<0.05) (92,93). However, this finding was based on less reliable data on diagnosis type from births occurring during the period before screening was widely available; no significant difference in survival between those detected through screening and those detected by symptoms other than meconium ileus was identified among those who received a diagnosis after 1986 (92,93).The survival curves were almost identical between the screened and symptomatic-detection post-1986 diagnosis groups through age 14 years. The analysis does not provide evidence of improved child survival through screening. Because newborn screening has been demonstrated to predict nutritional status, an indirect assessment of the effect of screening on survival can be derived from observational studies of the association of nutritional status with survival among children with CF (101). One multivariate analysis of CFNPR data determined that children with weight-for-age less than the fifth percentile between ages 1--5 years were four times more likely to die over the ensuing 8 years than patients with weight-for-age >50th percentile (95% CI = 2.1--7.3) (105). Height-for-age, the outcome most consistently associated with newborn screening in the Wisconsin trial as well as observational studies, has also been linked with mortality risk in CF. One analysis of CFNPR data during 1980--1993 indicated that male children aged 7 years with height-for-age below the fifth percentile had an unadjusted relative risk of dying during childhood and adolescence of 6.3 (95% CI = 2.1--16.7) (96). A multivariate analysis of adolescents and adults with CF in England also determined that height was strongly predictive of subsequent mortality (p<0.0001) (102). Cognitive Function The 1997 workshop recommended that data be collected to analyze cognitive outcomes among children with CF classified by age of detection to determine whether advantages in early growth might have lasting neurodevelopmental consequences (5). To this end, cognitive assessments were performed at ages 7--17 years on 89 children in the Wisconsin study for whom consent could be obtained, including 67 children without meconium ileus and with no missing data (100). Cognitive function was assessed with the Test of Cognitive Skills, 2nd Edition, which was used to generate the Cognitive Skills Index (CSI) (mean = 100; SD = 16), a validated measure of general intelligence equivalent to an IQ (106). Overall, the screened group had a 5-point higher mean CSI score relative to the control group, which is equivalent to one third SD, controlling for confounding variables. This finding was not statistically significant. No association was identified with any anthropometric indicator, including head circumference, height, and weight (100). A larger and statistically significant CSI differential was observed among the approximately 50% of children in the Wisconsin RCT who had vitamin E deficiency at time of diagnosis (100). The 17 control group subjects who had low alpha-tocopherol levels at time of diagnosis had adjusted CSI scores 12.5 points lower than scores for the 17 screened subjects who also had vitamin E deficiency at diagnosis (p<0.05). Differences in cognitive ability of this magnitude are perceptible to patients; a reduction of 1 SD in cognitive scores is associated with a decrement in reported HRQoL (107). Health-Related Quality of Life Health-related quality of life (HRQoL) is a common measure for studies of clinical utility. The 1997 workshop recommended that HRQoL among children and adolescents with CF be assessed to determine whether early intervention resulting from newborn screening leads to better outcomes from the person's own perspective (5). HRQoL can be assessed by using either generic instruments or disease-specific instruments; the majority of studies of HRQoL have used generic instruments (e.g., the Quality of Well-Being [QWB] Scale or Child Health Questionnaire [CHQ]). Studies that used generic HRQoL instruments with persons with CF have reported that HRQoL is negatively associated with recent morbidity (e.g., number of respiratory infections, concurrent medical conditions, and daily medications) but appears to be unrelated to nutritional status or lung function (108,109). In response to the 1997 workshop recommendation, a follow-up study of HRQoL was conducted among a subset of 89 participants in the Wisconsin RCT assessed at ages 7--17 years. Results using the CHQ indicate that HRQoL did not differ between the screened and control groups (107). The best predictor of lower HRQoL in one of the regression models was the number of recent hospitalizations, consistent with previous studies (108,109). Because the generic HRQoL instrument used in this study is reported to have limited sensitivity compared with disease-specific instruments for CF in measuring differences in health states (110), a more sensitive disease-specific instrument might have revealed between-group differences in HRQoL. Another limitation of the HRQoL study is that it was conducted long after diagnosis among respondents of different ages. More research on HRQoL among younger children with CF is warranted to assess the potential qualitative benefits of newborn screening. Cross-sectional assessments of HRQoL among children who already have received a diagnosis of CF cannot capture adverse effects on quality of life occurring during the period before diagnosis. For example, enteric-coated pancreatic enzyme therapy has been demonstrated to reduce the frequency of abdominal pain among older children, adolescents, and adults (111). By allowing for earlier initiation of enzyme therapy, newborn screening could lead to improvements in HRQoL among the subset of infants with gastrointestinal symptoms during the interval before initiation of therapy prompted by symptomatic diagnosis. Hospitalizations and Burden of Care As noted previously, studies among persons with CF have indicated that severe symptoms requiring multiple medications and hospitalizations are associated with statistically significant decrements in HRQoL (107,108,110). Because hospitalizations and prescription medications comprise the bulk of medical costs for persons with CF (112), any intervention that leads to reduced hospitalizations and concurrent pharmacologic treatments can substantially reduce costs to the health-care system and improve the well-being of affected children and their families. Evidence relating newborn screening to the cost and burden of hospitalizations or medications is suggestive of benefit for these patient-oriented outcomes. The U.K. RCT data indicate that children in the screened cohort spent a statistically significantly shorter amount of time in the hospital during the first year of life than those in the unscreened cohort (mean [SD] = 19.2 [42.9] and 27.0 [22.7] days, respectively) (p<0.01). The Wisconsin RCT has not confirmed a reduction in hospitalizations; 51% of persons in the screened group had a recorded hospitalization at any age, compared with 34% of the control group (69). The hospitalization rate for the unscreened group in Wisconsin was lower than expected and might reflect underascertainment of data on hospitalizations. An earlier analysis reported higher rates of hospitalization within the screened group controlling for CF center, although the differences were not statistically significant (76). Four observational studies have reported that diagnosis resulting from newborn screening is associated with fewer hospitalizations among young children with CF. In Australia, the average number of hospital days in the first 2 years of life was 27 for the unscreened cohort and 4 for the screened cohort (80). In France, the cumulative frequency of hospitalizations for children with CF is reported to have been significantly lower in a screened cohort than in a neighboring unscreened cohort (49% and 86%, respectively; p<0.001) (87). An analysis of CFNPR data from 2000--2002 on infants aged <12 months who received a diagnosis of CF reported that 22% of infants who received a diagnosis through newborn screening were hospitalized during the year of diagnosis, compared with 64% of infants who received a diagnosis on the basis of clinical symptoms (27). An earlier study from Colorado (113) reported an even larger differential, but that analysis was restricted to hospitalizations occurring before diagnosis, which is less meaningful. According to one study (114), antibiotic prophylaxis against infections with Staphylococcus aureus reduced hospitalizations among young children with CF detected through newborn screening, but conclusive evidence is lacking. One RCT conducted in East Anglia during 1985--1989 randomly selected 38 infants identified with CF through newborn screening to receive either continuous antibiotic prophylaxis with flucloxacillin or episodic treatment (114). During the second year of life, mean hospitalization admission rates in the prophylaxis group were one fourth as high, and mean duration of hospital stay was one third as high (p<0.01). Two RCTs not restricted to infants detected by screening (78,115) and using different antibiotics did not find a preventive effect of prophylaxis (116). One RCT identified an apparent risk of elevated P. aeruginosa isolation after 4 years of continual prophylaxis (115). Certain U.K. centers that use antistaphyloccocal prophylaxis with aggressive treatment regimens to eradicate P. aeruginosa infections report promising results in terms of improved outcomes (42,43). Evidence that early detection by screening might reduce the number of medications administered, especially more invasive medications, is more limited. In the French study, no difference between the two cohorts was observed in the frequency of intravenous antibiotic therapy. However, the screened cohort was approximately half as likely to use inhaled antibiotics, a clinically important (albeit not statistically significant) difference (87). An abstract from a CF care center in Southampton, England, reported that those receiving a diagnosis at age <2 months had statistically significantly lower rates of using intravenous antibiotics than those who received a diagnosis at age >2 months (0% and 19%, respectively), regular nebulized antibiotics (57% and 95%, respectively), and regular inhaled corticosteroids (36% and 85%, respectively) (all p<0.05) (117). UKCFD data indicate a statistically significant lower frequency of use of intensive therapy among children whose CF was detected by screening compared with those whose CF was detected by clinical diagnosis (91). Among children aged 1--3 years, those whose CF was detected through screening were only half as likely (22% versus 44%) to receive nebulized antibiotics, mucolytics, or corticosteroids. Among children aged 4--6 years, the comparable percentages were 41% and 60%, respectively; p<0.05). By ages 7--9 years, the majority of children in both groups (55% and 67%, respectively) were receiving such therapies. The lower use of therapies in the screened group might indicate that a main benefit of screening is the ability to achieve the same health outcomes with less burden of treatment on children and families and less cost to the health-care system (91,117). Benefits to FamiliesChildren with CF who receive a diagnosis on the basis of clinical symptoms rather than screening usually experience a series of diagnostic tests and treatments before a conclusive diagnosis of CF is made (29). A 1991 Colorado study determined that 86% of children with CF who were not detected through screening received an incorrect or incomplete diagnosis before a correct diagnosis of CF, and all had undergone hospitalization (113). On average, 15 months were reported to have elapsed between the onset of symptoms and a definitive diagnosis of CF. The typical delay from symptoms to diagnosis is somewhat shorter, with the median age of diagnosis for those receiving a diagnosis before age 20 years on the basis of symptoms reported to be 14.5 months (27). The adverse effects of delayed diagnosis on families of children with CF have been extensively studied (118). Parents of children detected with CF on the basis of clinical symptoms are more likely to report experiencing personal distress (119); anxiety (118); and negative feelings toward, and distrust of, medical professionals (120,121). In the United States, a synthesis of family experiences with delayed diagnosis includes anxiety, stress, frustration, self-doubt, and emotional trauma (29). The possibility of sparing parents the experience of emotional pain as a result of delayed diagnosis might be an important benefit of newborn screening. However, receiving a diagnosis of CF usually causes anxiety, regardless of mode or age of diagnosis, and parents of children with CF continue to experience high levels of stress no matter how the diagnosis was originally established (113,122). Another potential benefit to parents from a diagnosis of CF by newborn screening is the ability to make informed decisions related to further childbearing, because the diagnosis might occur 1 year earlier on average compared with conventional diagnosis (0.5 and 14.5 months, respectively) (27). Six studies in Europe and Australia indicated that the majority of couples with positive CF newborn screening results use that information in their childbearing decisions (123). Limited evidence from two U.S. studies suggests less reproductive effect of newborn screening (113,124), which might reflect differences in genetic counseling or cultural attitudes (125). Evidence for Risks of HarmPotential harms from newborn screening can occur both to children with CF and their families and to families of children who have a false-positive newborn screen. On the basis of previous experience with genetic screening programs (1,126,127), certain psychosocial risks were anticipated, notably parental anxiety and disturbance of the parent-child bond. These risks are related to the communication process and the experience of families being informed about screening results (14). Appropriate resources for diagnostic evaluations, treatment for infants with CF, and counseling for families might help mitigate these risks (128--130). Ongoing quality-assurance programs built into the screening system are needed to provide accurate data on risks. Risks to Infants Affected with CF and Their FamiliesAltered Parent-Child Relationships Before the introduction of newborn screening for CF, the possibility that screening could have an adverse effect on the bonding process between new parents and their infants had been hypothesized (127). Studies of families of children with CF have investigated the hypothesis, and none has determined this to be a problem. For example, a study of 58 families of Australian children with CF, half of whom were detected through newborn screening, did not find evidence of altered maternal behavior in response to a diagnosis of CF through newborn screening (119). A study from Wales in 1999 of nine mothers of children with CF detected through newborn screening and 82 mothers from the general population identified no evidence of greater rejection or overprotection in the CF group (131). Although the number of families studied is limited, these reports suggest that early identification of CF through newborn screening does not adversely alter parent-child relationships. Person-to-Person Transmission of Infectious Agents The Wisconsin RCT documented earlier acquisition of P. aeruginosa among infants detected through screening who were examined at a CF center housed in a 90-year-old building with a 110-square-foot waiting room in which screened infants were not isolated from older children with CF, many of whom had lung infections (28,30).¶¶ Children in the screened group who were examined at the first center acquired P. aeruginosa at a median age of 1.0 years, compared with a median of 5.6 years for those examined at the other center (76). Evidence from Australia documenting acquisition of P. aeruginosa from other persons with CF cared for at the same center supports the hypothesis that this is a concern in the treatment of infants and young children with CF (132). Centers caring for children with CF should address this problem, whether CF is detected clinically or through newborn screening. The Wisconsin evidence indicates that person-to-person transmission of P. aeruginosa is not an inherent risk of newborn screening and can be avoided. It does not appear to be common among children with CF detected by newborn screening in the United States (27). A study of 180 children in Colorado detected by newborn screening identified a median age of acquisition of P. aeruginosa of 8.1 years (133). In 2003, CFF issued new recommendations to minimize the risk of person-to-person transmission of infectious agents, including segregating asymptomatic patients from those with established disease (134). Studies to evaluate the efficacy and use of infection-control policies should be conducted. Risks to Infants with False-Positive CF Screens and Their FamiliesThe anticipation of psychosocial risks to parents whose infants have a false-positive CF screening result was based on previous experiences with newborn screening for PKU (135) and with other benign conditions (e.g., hyperbilirubinemia and heart murmurs) that resulted in anxiety and overprotectiveness among certain families (136). With a simple IRT/DNA approach, all children with positive screens either have CF or are CF carriers (62), although this is not the case in programs that also refer children with highly elevated IRT and no detected mutations. Psychological Distress from Screening Parents of children who screen positive for CF, the majority of whom do not have CF, often report feeling distress and anxiety. One study from Wisconsin of 104 parents whose infants had elevated IRT measures indicated that parents surveyed while waiting for results of sweat tests reported feelings of concern (96%), depression (77%), and shock (76%) (120). A minority of families of children with false-positive screen results reported lingering anxiety even after being informed that their child did not have CF. A study in Wisconsin of 106 parents whose infant had positive screens under the IRT screening algorithm and 63 parents whose infants screened positive under the IRT/DNA (single-mutation) algorithm (none of whom had CF) determined that, when interviewed 1 year after screening, 7% and 10%, respectively, reported that they worried about the results once a week or more (124). Misunderstanding of Carrier Status The information that a child with a positive screen is a carrier, that at least one of the parents is a carrier, and that the parents are at increased risk of having a child with CF, is complex to convey. A Wisconsin study of 63 families with positive IRT/DNA test results reported that 6% of families thought that being a carrier could cause illness (124). Education and genetic counseling might improve understanding, but a second study from Wisconsin of 138 families conducted after CF screening had become part of the state newborn screening program indicated that only two thirds of parents received genetic counseling (i.e., counseling from a physician, nurse, or genetic counselor) (137). In this cohort, 88% understood that their child was a carrier, but not all had accurate understanding of the implications; 11% either believed that their child would have CF subsequently or did not know whether that could happen (137). Approximately half of parents reported feeling confused, and only one fourth of families reported feeling no anxiety about their child's carrier status. Less than half of the parents, regardless of whether they reported receiving genetic counseling, understood that they were at increased risk of having a child with CF. These findings underscore the need for comprehensive communication plans, research about effective risk-communication strategies, and assessments of parental understanding of routine screening programs (128). Other Implications to Families of Carrier Identification The provision of information about CF carrier status has raised concerns about potential stigmatization and discrimination (138), although subsequent studies have not yielded evidence of such adverse effects. In addition, certain persons have reported feeling uncomfortable with the implications of contacting other family members about CF carrier status (131). States screening newborns for SCD*** have long experience in reporting sickle cell trait, which poses similar challenges in communicating complex genetic information to families (139). Despite these concerns, studies indicate that the majority of parents of carriers detected by screening consider carrier identification to be a useful by-product of newborn screening for CF. In Wisconsin, 14% of parents of carriers in one sample reported that they did not think newborn screening for CF should be conducted, whereas two thirds of families strongly agreed that they were better informed by knowing their infant's carrier status and would not have preferred that it had remained unknown (137). In Massachusetts, where newborn screening for CF is optional, preliminary analysis of data on linked births indicated that 82 of 83 families who had an infant with a positive screen agreed to newborn screening for CF for their subsequent infants (140). Also, a study of 10 carrier families in Wales reported that all were in favor of newborn screening for CF (131). In Massachusetts, one genetic counseling program reported that 75% of families of infants identified as carriers requested carrier testing for both parents; among the five families in which both parents were identified as CF carriers, an older sibling in one family was subsequently identified as having CF (141). Risks to Health-Care SystemWhenever a condition is added to a newborn screening panel, potential risks can occur as a result of adding responsibilities to the health-care system (129). First, infants with false-negative results might experience a delay in diagnosis. Second, overburdening the system with children who are false-positives could distract primary-care providers and specialists and complicate addressing the diagnostic and psychosocial needs of families. These risks can be minimized by optimizing the screening protocol to minimize false-negative and false-positive results and by ensuring that adequate resources are available. These two risks emphasize the importance of collaboration between newborn screening programs and health-care providers in planning and implementing a screening program. Finally, screening always has an opportunity cost, which consists of the diversion of health-care system resources away from other potential uses. Failure To Diagnose CF The American Academy of Pediatrics (AAP) urges health-care providers to be vigilant to detect disorders included in newborn screening programs (60,142). This is of particular importance for CF, because newborn screening for CF results in more false-negatives than are documented for other established newborn screening tests (range of sensitivity in routine programs: 93%--98%) (Table 2). Providers in states that screen for CF need to remain vigilant to detect CF among children with clinical symptoms. In particular, providers should consider the possibility of CF among children with meconium ileus, children of non-European ancestry who might carry mutations not included in mutation panels used by screening programs, and children who were born in a state without screening. A potential drop in requisitions by primary-care providers for sweat tests after newborn screening is implemented could result from physicians not ordering sweat testing on suspicion of CF even when warranted, thereby risking delayed diagnosis, or a reduction in unnecessary ordering of sweat tests with a low degree of suspicion of CF. In Wisconsin, the number of sweat tests requisitioned to test for CF is reported to have decreased 50% statewide after screening was implemented (68). A reduction of at least one third in the aggregate number of sweat tests ordered after introduction of newborn screening for CF has been reported from South Australia (Enzo Ranieri, M.D., Women's and Children's Hospital, Adelaide, South Australia, personal communication, May 4, 2004). Another approach to assessing whether changes in provider behavior might contribute to delayed diagnosis among those not detected by screening is to compare the median age at diagnosis among children identified through newborn screening with those identified through clinical diagnosis in a state with universal screening. Unpublished data indicate that all children with CF who have received a clinical diagnosis since the advent of routine screening in Wisconsin (in 1994) and in Massachusetts (in 1999) received a diagnosis by age 12 months (median age at diagnosis: <4 months) (Gary Hoffman, Wisconsin State Laboratory of Hygiene, Madison; Anne Marie Comeau, Ph.D., New England Newborn Screening Program, Jamaica Plain, Massachusetts, personal communications, April 6, 2004). In Colorado, the median age of diagnosis for 14 children with CF who screened negative is <8 months (Marci Sontag, Ph.D., The Children's Hospital, Denver, Colorado, personal communication, April 8, 2004), which is below the national average of 14.5 months for diagnoses based on clinical symptoms (27). Thus, no evidence exists that providers have changed behavior in states with newborn screening for CF. Effect of False-Positives The data indicate that the balance between numbers of screen-positives and cases of diagnosed CF is favorable compared with established newborn screening tests as long as an IRT/DNA screening algorithm is used (67). An IRT-repeat IRT protocol can yield similarly low numbers of false-positives if second specimens are routinely collected from all infants (18). Screening programs that use an IRT-repeat IRT protocol with a screen-positive report based on a single specimen can expect to generate higher numbers of screen-positives relative to true-positive cases. Every child who screens positive for CF should be referred to a specialty center for a diagnostic analysis. Health-care providers in Massachusetts have treated screen-positive results with due diligence, with >95% of infants with positive screens completing sweat testing (67). A concern relating to false-positives is that an increase in the number of referrals for sweat tests or genetic counseling could overwhelm CF care centers, thereby adversely affecting care received by persons with CF. Although CF care centers will initially need to be prepared to handle a higher volume of sweat tests, the increase might be modest and transient, depending on the screening algorithm used in a state. One CF care center in Massachusetts reported a 10% increase in numbers of sweat tests requisitioned after the introduction of screening (Richard B. Parad, M.D., The Children's Hospital, Boston, Massachusetts, personal communication, April 15, 2004). The availability of resources for providing genetic counseling for families of carriers as well as sweat testing should be addressed before implementation of newborn screening (140). Minimizing RisksAll newborn screening tests (including tests for CF) pose potential risks of harm that need to be minimized through careful attention to concerns of communicating screening results and genetic risks and coordination of follow-up among diagnostic centers, primary-care physicians, genetic counselors, specialists, and families (4,129,143). Screening programs might minimize psychosocial risks by acting to minimize time delays between informing parents of positive results and providing sweat tests; improve the type and nature of information given to parents about the meaning of an initial positive screening result, diagnosis of CF, or identification as a carrier; and provide psychological support for the families of infants with CF and for CF carriers who have ongoing concerns (118). If providers were to educate parents in a relaxed manner before labor and delivery, a positive screening result might be met with greater willingness to obtain follow-up and with less worry about the meaning of a positive test (144,145). Newborn screening information provided to parents at the time the heel stick is taken, soon after birth in the United States, might be poorly assimilated and not contribute to effective understanding, and parents clearly prefer to receive this information before testing (142,146). Better parental education about newborn screening for all disorders is vitally important and ideally should occur in the prenatal setting (144--148). Information should be provided in multiple formats (e.g., easy-to-read pamphlets and videos) (149). Obtaining parental permission might facilitate parental engagement and education. Parental consent for newborn screening for PKU and other disorders has been debated since 1978 (150). Forty-eight states mandate newborn screening because treatment begun soon after birth has been demonstrated to prevent irreversible, devastating outcomes (e.g., death or severe intellectual disability for certain disorders). When the balance between benefits and risks for newborn screening tests is not as dramatic as it is for PKU, the argument for parental consent becomes more compelling (14,151). Consent includes a spectrum of approaches, ranging from providing information and obtaining verbal agreement to written documentation of consent with a signature (152). A report from an ad hoc Newborn Screening Task Force convened in 1999 recommended that parents should be informed and be able to refuse testing, although documentation of consent should not be necessary (142). AAP has recommended that states evaluate an informed consent process for newborn screening tests to foster parental education and promote informed responses to test results (144). Consent does not preclude providers from making strong recommendations in favor of testing, and if the risk is limited and clinical recommendations have been made to offer a test, the approach to consent can be simple (153). In Massachusetts, where newborn screening for CF requires oral consent from a parent (i.e., a check box on the newborn screening card indicates refusal), >98% of parents consent to CF newborn screening (17). Weighing Risks and BenefitsEvidence of BenefitsHealth Outcomes The 2003 workshop reviewed the U.S. and international peer-reviewed medical literature to assess the efficacy of newborn screening for CF in light of new data appearing since the 1997 workshop. The results support the efficacy of newborn screening in reducing morbidity from CF (Table 4). In particular, the benefits of improved growth are now more clearly established than in 1997, and the implications of growth retardation for other clinical outcomes in CF are better understood. In addition, benefits in terms of improved patient-oriented outcomes, including cognitive outcomes, hospitalizations, and survival, have been reported in recently published studies. Unlike certain conditions for which universal newborn screening is mandated (e.g., PKU, congenital hypothyroidism, and galactosemia), CF is not associated with severe intellectual disability. For this reason, improvement in cognitive scores, although meaningful, is not equivalent to prevention of mental retardation. Results of studies do not demonstrate clear benefits on other important measures, including HRQoL and pulmonary outcomes. No information is available on cost-effectiveness, although partial cost analyses have been published. Finally, newer research and the experience of additional cohorts with screening have tended to alleviate concerns about risks of psychosocial harm among children with CF but have continued to find evidence of anxiety or misunderstanding among parents whose children have false-positives results. Growth and nutrition. As was true in 1997, the most clearly defined benefit from newborn screening is improved growth of affected children. In particular, the Wisconsin RCT provides strong evidence of long-term improvements in height-for-age and reductions in chronic malnutrition. A second analysis published in 2001 strengthened this finding by demonstrating that the height advantage in the screened group persisted through age >10 years and by including growth measurements for a subset of the control group that had previously been excluded (77). In the SORT framework, a finding from a single high-quality RCT that an intervention substantially improves a patient-oriented outcome is considered to provide Level-1 evidence (Box 2). As a disease-oriented outcome, evidence from an RCT is considered Level-3 evidence for recommendation of newborn screening for CF. However, if height-for-age below the normal range were considered a patient-oriented outcome, this would be considered Level-1 evidence. Studies in other countries also support improvements in height-for-age from screening for CF. In addition, anthropometric measures of nutritional status have been linked to reduced mortality among children and adults with CF independent of newborn screening, which suggests that early initiation of nutritional therapies after newborn screening might lead to improved long-term survival (101). Cognitive development. Newborn screening has also been demonstrated to influence neurodevelopment positively in a subset of children with CF. Results from the Wisconsin RCT indicate moderate, clinically significant improvements in general cognitive ability assessed in school-age children and adolescents among those with vitamin E deficiency at time of diagnosis (100). Assuming that a 12.5-point difference in cognitive ability is considered a patient-oriented outcome, this finding qualifies as Level-1 evidence in the SORT framework. The observed overall difference of 5 points in mean cognitive test scores between screened and control groups, which was not statistically significant, is comparable in magnitude to that associated with moderately elevated blood lead levels (154). Other CF follow-up studies should conduct cognitive assessments as well. Hospitalization. Studies indicate a substantial benefit of newborn screening in reducing the days of hospitalization for children with CF, including one RCT from the United Kingdom (78) and observational studies from Australia (80), France (87), the United Kingdom (91), and the United States (27). A preliminary analysis of retrospectively collected data from the Wisconsin RCT has not demonstrated this to be the case, which might be attributable to how the data were collected or to differences in treatment protocols. In addition to the potential for newborn screening for CF to reduce costs to health-care systems from hospitalizations, findings from Wisconsin and Australia indicate health-care savings from reduced ordering of sweat tests ordered by primary care providers. Survival. A number of observational studies and one RCT have reported Level-2 evidence of proportional reductions of >50% in mortality rates among children with CF diagnosed by newborn screening. Child mortality as a result of CF is low in absolute terms (44), and the majority of the findings from individual studies lack statistical significance. No studies have reported equal or worse survival among screened children, although the lack of negative findings might reflect publication bias. A meta-analysis of survival data might provide more robust estimates of the effect on survival of newborn screening for CF. In addition, the frequency of children with CF dying without having received a diagnosis can be studied through death records linked with stored residual blood spot specimens from programs not screening for CF. Pulmonary status. The effect of newborn screening on pulmonary status among persons with CF as measured by chest radiographs and lung function remains uncertain. Three observational studies, including two conducted in recent years, have demonstrated evidence of less severe lung damage among children identified by newborn screening, although no difference in lung function was demonstrated in two of the three studies (Table 4). One cohort study from Australia reported evidence of both better lung function and less lung damage among children with CF born after screening was implemented. Although the randomized Wisconsin trial identified no benefit for any pulmonary measure through age 16 years and a possible disadvantage in terms of chest radiographs, the study was weakened by probable person-to-person transmission of P. aeruginosa in one clinic that did not segregate patients, thus possibly contaminating the results for this cohort. In addition, the control group by chance had relatively more subjects with pancreatic insufficiency and milder symptoms. Even if the Wisconsin RCT had provided evidence of pulmonary benefit, this would be classified as Level-3 evidence in the SORT framework, because it is a disease-oriented outcome. HRQoL. Evidence of benefit is lacking for improved HRQoL as a patient-oriented outcome measure for the efficacy of newborn screening for CF. The Wisconsin study did not identify significant differences in HRQoL between the screened and control groups at age 7--14 years. The limited literature on HRQoL relevant to CF suggests that differences in HRQoL are related directly not to nutritional status or lung function but primarily to the frequency of pulmonary exacerbations and treatment interventions, including hospitalization. Distribution of benefits. For certain children detected through screening, the benefits are more substantial than for others. For example, greater growth is primarily of benefit to those who would have been below the normal range in the absence of screening. Similarly, the cognitive benefits from screening are concentrated among those children at risk for micronutrient deficiency. CF newborn screening is likely to be most helpful for that subgroup of affected children who do not receive a prompt diagnosis despite severe disease manifestations. Benefits to this subset of children through screening might not be reflected adequately in comparisons of mean outcomes. Similarly, although child mortality as a result of CF is low, early detection might save an infant from a rare life-threatening event (e.g., hyponatremic dehydration). However, persons with pancreatic sufficiency, who comprise <10% of persons with CF, might not benefit from newborn screening (4). Similarly, the 20% of persons with meconium ileus do not appear to benefit from newborn screening because they typically receive a diagnosis at birth. Benefits to Families In addition to health benefits for children, which are the primary justification for newborn screening for CF, newborn screening offers potential benefits to families. One such benefit is the elimination of the diagnostic odyssey. Further, certain families value the genetic risk information received from newborn screening for CF. Another benefit of carrier testing, at least in the early years of newborn screening implementation, is the identification of older siblings who are symptomatic but have not received a diagnosis of CF after a positive screen for CF in a newborn infant (140). Overall, studies report that the majority of families of children who either screen positive or receive a diagnosis of CF consider newborn screening for CF to be worthwhile. Risks of Harms and CostsThe potential benefits of newborn screening programs should be weighed against the potential harms and costs. Harm from person-to-person transmission, which is not inherent to earlier detection, can be avoided through involvement of newborn screening programs with CF care centers and adoption of infection control practices. The potential negative outcomes of screening include effects from false-positive results and the psychological and social effects of carrier identification for parents and children. Studies indicate that certain parents experience anxiety from false-positive results and confusion over the implications of CF carrier status. Screening programs should explore options for minimizing parental anxiety and misunderstanding. Concerns that should be addressed include better communication of screening results and provision of more comprehensive information about newborn screening. AAP has called for facilitating informed parental choices regarding newborn screening (142,144). Documentation of consent might not be necessary. The focus should be on providing thorough, easily understood information to parents about screening for CF and other conditions, especially before delivery, to reduce misunderstanding and provide parents with an opportunity to make informed choices, consistent with state laws. A full cost-effectiveness analysis of the costs and benefits of newborn screening has not been conducted. Partial cost analyses conducted in Wisconsin indicated that the majority of CF screening costs in that state were offset by savings from the reduction in the ordering of sweat tests (68,69). Other potential economic benefits are reductions in hospitalizations and medications for screened children that have been reported, although not in the Wisconsin study. Indeed, one of the major benefits of newborn screening for CF might be to reduce the level of treatments and costs needed to maintain comparable health status between screened and unscreened children (91,117). In the absence of formal economic evaluations, states should assess the economic benefits of newborn screening for CF compared with other alternative actions that might be foregone if resources were allocated to CF screening (e.g., adding another disorder to newborn screening panels). Both risk and costs might be influenced by decisions about testing methodology (e.g., IRT cutoffs and the numbers of mutations included). As with all screening programs, a tradeoff exists between sensitivity and specificity. Benefits can be maximized by identifying more children with CF (higher sensitivity), and risks can be reduced by decreasing the number of carriers detected (higher specificity). Detecting more children with CF by increasing the number of mutations tested for by using the IRT/DNA approach necessarily increases the number of carriers detected. The Wisconsin program has reported that switching from a single mutation to a 25-mutation panel raised screening costs by one third to one half but reduced the frequency of false-negatives by more than half (Table 2) (69). The potential for harms underscores the need for continual monitoring and evaluation of screening outcomes to ensure that children with CF benefit from screening and interventions. An example is an evaluation by the Colorado newborn screening program relative to P. aeruginosa acquisition (133). Moreover, data collection and analysis are needed to evaluate and optimize the performance of screening and diagnostic algorithms (17). Balance of Benefits and RisksOn the basis of the SORT taxonomy (Box 2) as applied in this report, the evidence of health benefits merits a B-level recommendation for newborn screening for CF. This is based on evidence for several patient-oriented outcomes. This recommendation includes consistent Level-2 evidence for improved child survival, although not all studies find statistically significant differences. Although Level-2 evidence of benefit in terms of reduced hospitalizations has also been reported from multiple studies, the Wisconsin RCT provided inconsistent findings. One high-quality RCT has yielded positive findings for two outcomes, growth and cognitive ability. Level-1 evidence for cognitive outcome supports a B-level recommendation. If impaired growth were classified as a patient-oriented outcome, Level-1 evidence from the Wisconsin RCT and Level-2 evidence from several observational studies would also provide support for a B-level recommendation. On the basis of a preponderance of evidence, the health benefits to children with CF outweigh the risk of harm and justify screening for CF. The complex policy decision as to whether to adopt screening also requires consideration of costs, resources, and priorities (14). As a result, CDC believes that including screening for CF in state newborn screening programs is justified. The evidence of clinical benefits from newborn screening for CF is based on an extensive body of research, including two RCTs and multiple prospective cohort studies. Indeed, the CF research experience could be a model for developing an evidence base for use in assessing the risks and benefits of other newborn screening tests (144). The net balance of benefits and risks is contingent on how newborn screening for CF is implemented (129). Consequently, newborn screening programs for CF, if initiated, should be of high quality and carefully monitored to ensure consistent quality and effectiveness. CF screening programs are complex and should be developed in a deliberate fashion with attention to the experience of existing programs. Benefits are likely to be maximized if children have access to state-of-the-art therapy and follow-up with experienced professionals. Adoption of newborn screening for CF should be accompanied by an implementation planning process involving specialized CF care centers and specialists in risk communication, including genetic counselors. An implementation plan should ensure that adequate personnel and other resources required for the accurate diagnosis and clinical management of young children with CF and the psychosocial and genetic counseling needs of families with screen-positive infants are made available to ensure effective and equitable access to services. RecommendationsThe magnitude of the health benefits from screening for CF is sufficient that states should consider including routine newborn screening for CF in conjunction with systems to ensure access to high-quality care.
AcknowledgmentsThe preparers acknowledge the contributions made by Denise Green, M.P.H., of CDC's National Center on Birth Defects and Developmental Disabilities (NCBDDD) in coordinating the logistical arrangements for both the 2003 workshop and the preparation of this report. CDC staff who reviewed drafts of this report include Pamela Costa, M.S., Owen Devine, Ph.D., Richard Olney, M.D., NCBDDD, and Muin J. Khoury, M.D., Mary Lou Lindegren, M.D., and Linda Bradley, Ph.D., Office of Genomics and Disease Prevention. Other workshop participants who offered comments were Michael Glass, M.S., Newborn Screening, Washington State Department of Health; Michael J. Rock, M.D., Cystic Fibrosis Center, University of Wisconsin, Madison; Lainie Friedman Ross, M.D., University of Chicago, Illinois; Marci Sontag, Ph.D., The Children's Hospital, Denver, Colorado. Gurvaneet Randhawa, M.D., and Elizabeth Edgerton, M.D., from the Agency for Healthcare Research and Quality, Rockville, Maryland, also provided helpful comments and suggestions. We also thank the members of our external review group for their helpful input: Preston W. Campbell III, M.D., Cystic Fibrosis Foundation, Bethesda, Maryland; Philip M. Farrell, M.D., University of Wisconsin Medical School, Madison; Kenneth S. Fink, M.D., Agency for Healthcare Research and Quality, Rockville, Maryland; Michele A. Lloyd Puryear, M.D., Health Resources and Services Administration, Rockville, Maryland; Richard B. Parad, M.D., New England Newborn Screening Program, University of Massachusetts Medical School, and Divisions of Newborn Medicine and Pulmonary Medicine, The Children's Hospital, Boston. References
* The number of persons with CF at birth relative to the size of the birth cohort, even though they might not receive a diagnosis until later, because CF is a genetic condition. † On the basis of data from states that reported confirmed results; however, reporting is incomplete (21). § The sensitivity for carrier screening is lower because, to be identified as a carrier couple, both parents must have a mutation included in the mutation panel. ¶ Refers to persons with a positive screening result who do not receive a diagnosis of disease. A positive screening result might provide useful information even absent a diagnosis. Infants with a positive screening result for CF identified using an IRT/DNA algorithm who do not receive a diagnosis of CF are carriers with one CFTR mutation (17). ** The Australian cross-sectional analysis identified significant differences in height-for-age (p<0.05) but not in weight-for-age; a longitudinal analysis of a subset of observations identified significant differences in weight-for-age (p<0.05) but not in height-for-age (81). †† One study reported a possible association of early asymptomatic detection and lung function among a subset of children born during 1987--1990, but ascertainment bias was a problem, because only children who received a diagnosis by the end of 1990 were included (26). §§ Four deaths not related to CF were recorded among screened children with CF in Brittany, three of which occurred neonatally. Infants with CF who died from such causes would not have received a diagnosis of CF in the absence of screening. In addition, one child with CF in Brittany died from causes associated with Down syndrome (87). ¶¶ Person-to-person transmission of P. aeruginosa in the Wisconsin study was not documented through microbiologic analysis. *** As of August 2004, all but two states routinely screened newborns for hemoglobinopathies (9).
Terms and Abbreviations Used in This Report
|
| AAP American Academy of Pediatrics | IRT immunoreactive trypsinogen |
| ACMG American College of Medical Genetics | IRT/DNA screening protocol/algorithm in which a blood spot specimen with an elevated IRT value is tested for the presence of CFTR mutations |
| allele alternative form of a gene that exists at a specific gene location (locus) on a chromosome | IRT-repeat IRT screening protocol/algorithm in which an infant with an elevated IRT on an initial blood spot specimen has a second specimen collected that is analyzed for elevated IRT |
| AMP adenosine-5'-monophosphate | lung function measures of flow of air out of the lungs and how much air the lungs can hold |
| analyte substance measured by a laboratory test | meconium ileus an intestinal obstruction present at birth, due to abnormally thick meconium that blocks the passage of stool out of the ileum and into the colon |
| analytic validity ability of a test to accurately and reliably measure a specific analyte or identify a mutation of interest, including analytic sensitivity and specificity | mutation a permanent heritable change in the molecular sequence of a gene |
| anthropometry measurement of the size and proportions of the human body and its parts | newborn screening public health service for the early detection of treatable disorders on the basis of laboratory analysis of dried blood spot specimens collected from newborn infants |
| autosomal recessive allele located on one of the autosomal chromosomes inherited from each parent; presence of two similar alleles is required to cause a particular clinical finding or disorder | nonclassic CF mild form of CF in which patients retain CFTR and pancreatic function; organ systems might be affected to a lesser degree than in classic CF |
| birth prevalence number of cases of a specified condition in a birth cohort relative to the number of births | pancreatic insufficiency absence or virtual absence of pancreatic enzyme activity, resulting in reduced ability to digest and absorb fat and fat-soluble vitamins |
| carrier person who has just one copy of a recessive disease-causing allele; does not have the disease in question | phenotype clinical presentation or expression of a genotype |
| CF Cystic fibrosis | pilocarpine iontophoresis activation of sweat glands on localized area of skin by a positive electrode moistened with pilocarpine nitrate; also known as sweat testing |
| CFF Cystic Fibrosis Foundation | PKU phenylketonuria |
| CFNPR Cystic Fibrosis Foundation National Patient Registry | positive predictive value the likelihood that a person with a positive test result actually has the disease |
| CFTR Cystic fibrosis transmembrane conductance regulator protein | QWB quality of well-being; instrument used to measure HRQoL |
| CFTR gene that regulates expression of the CFTR protein | RCT randomized controlled trial |
| CHQ Child Health Questionnaire; instrument used to measure HRQoL | RSV respiratory syncytial virus |
| CI confidence interval | SCD sickle cell disease |
| clinical utility net balance of outcomes (risks, benefits, and costs) demonstrated to result from screening, including interventions following diagnosis, in comparison to the natural history of the disorder in the absence of screening | screen-positive person whose screening result is positive for a marker of a disease |
| clinical validity ability of a test or protocol to accurately and reliably identify individuals who either have or will have the disorder or phenotype of interest | screening algorithm clinical decision pathway involving a series of screening tests and decision points whether to refer for further testing |
| CSI Cognitive Skills Index; measure of general cognitive ability | SD standard deviation |
| failure to thrive restricted growth in weight and length in an infant or toddler with unspecified cause | sensitivity frequency with which a test yields a positive result when the disease marker or disease in question is actually present in the person being tested |
| false-negative negative screening result reported for a person who actually has the disease of interest | Shwachman score clinical score for CF with four sub-scores: physical exam, nutrition, activity, and chest radiography |
| false-positive positive screening result reported for a person who does not have the disease of interest | SORT Strength of Recommendation Taxonomy; a classification of types of evidence for recommending clinical interventions |
| FEV1 forced expiratory volume in 1 second, a measure of lung function | specificity frequency with which a test yields a negative result when the disease marker or disease in question is not present in the person being tested |
| genotype a person's genetic makeup, specifically the alleles present at specific gene loci | spirometry method of quantifying flow of air out of lungs by using mouthpiece attached to a recording device |
| height-for-age a person's height relative to the heights of others of the same age and sex in a reference population | sweat testing collection and analysis of sweat electrolytes to diagnose CF on the basis of elevated sweat chloride levels |
| homozygote a person with two copies of the same allele of a gene | UKCFD United Kingdom Cystic Fibrosis Database |
| HRQoL health-related quality of life | USPSTF United States Preventive Services Task Force |
| intent-to-treat analysis that classifies persons according to the treatment that they were supposed to have received rather than what they actually experienced | Z-score number of standard deviations by which a measurement differs from the mean of the reference distribution |
| IQR interquartile range (i.e., the range between the 25th--75th percentiles) |
Cystic Fibrosis Meeting Participants
November 2003
Frank J. Accurso, M.D., University of Colorado, Denver, Colorado; Ran D. Anbar, M.D., SUNY Upstate Medical University Hospital, Syracuse, New York; Mary Ann Baily, Ph.D., The Hastings Center, Garrison, New York; Robert J. Beall, Ph.D., Cystic Fibrosis Foundation, Bethesda, Maryland; Stanton L. Berberich, Ph.D., University of Iowa Hygienic Laboratory, Iowa City, Iowa; Drucy Borowitz, M.D., State University of New York, Buffalo, New York; Jeffrey R. Botkin, M.D., University of Utah, Salt Lake City, Utah; C. Michael Bowman, Ph.D., M.D., Medical University of South Carolina, Charleston, South Carolina; Coleen A. Boyle, Ph.D., CDC, Atlanta, Georgia; Don Bradley, Ph.D., University Hospital of Wales, Heath Park, Cardiff, United Kingdom; Linda Bradley, Ph.D., CDC, Atlanta, Georgia; Edward Brann, M.D., CDC, Atlanta, Georgia; Peter Briss, M.D., CDC, Atlanta, Georgia; Preston W. Campbell III, M.D., Cystic Fibrosis Foundation, Bethesda, Maryland; Ellen Wright Clayton, M.D., J.D., Vanderbilt University, Nashville, Tennessee; Richard C. Cohen, M.D., Kaiser Permanente Northwest Region, Portland, Oregon; Anne Marie Comeau, Ph.D., University of Massachusetts Medical School, Jamaica Plain, Massachusetts; Krista Stimson Crider, Ph.D., CDC, Atlanta, Georgia; George Cunningham, M.D., California Department of Health Services, Richmond, California; Jeannette E. Dankert-Roelse, M.D., Ph.D., Vrije Universiteit Medical Centre, Amsterdam, The Netherlands; George W. Delavan, M.D., Utah Department of Health, Salt Lake City, Utah; Franklin Desposito, M.D., New Jersey Medical School, Newark, New Jersey; Owen J. Devine, Ph.D., CDC, Atlanta, Georgia; Jean-Louis Dhondt, M.D., Ph.D., Hospital St. Philibert, Lomme, France; James P. Dillard, Ph.D., University of Wisconsin, Madison, Wisconsin; Henry L. Dorkin, M.D., Massachusetts General Hospital, Boston, Massachusetts; Marie Earley, Ph.D., CDC, Atlanta, Georgia; Philip M. Farrell, M.D., Ph.D., University of Wisconsin Medical School, Madison, Wisconsin; Andrew Faucett, M.S., CDC, Atlanta, Georgia; Paul Fernhoff, M.D., Emory University, Atlanta, Georgia; Kenneth S. Fink, M.D., Agency for Healthcare Research and Quality, Rockville, Maryland; Norman Fost, M.D., University of Wisconsin Medical School, Madison, Wisconsin; Dennis Freer, Ph.D., Pediatrix Screening, Bridgeville, Pennsylvania; Patricia Furlong, MS, Parent Project Muscular Dystrophy, Middletown, Ohio; Michael Glass, M.S., Washington State Department of Health, Shoreline, Washington; Denise Green, M.P.H., CDC, Atlanta, Georgia; Nancy S. Green, M.D., March of Dimes, White Plains, New York; Scott D. Grosse, Ph.D., CDC, Atlanta, Georgia; Keith B. Hammond, M.S., University of Colorado School of Medicine, Denver, Colorado; W. Harry Hannon, Ph.D., CDC, Atlanta, Georgia; Mark Helfand, M.D., Oregon Health Sciences University, Portland, Oregon; Gary Hoffman, Wisconsin State Laboratory of Hygiene, Madison, Wisconsin; Niels Hoiby, D.M.S., Rigshospitalet, Copenhagen, Denmark; R. Rodney Howell, M.D., National Institutes of Health, Bethesda, Maryland; Michelle Howenstine, M.D., Indiana University School of Medicine, Indianapolis, Indiana; Robert A. Kaslovsky, M.D., Albany Medical College, Albany, New York; Aileen Kenneson, Ph.D., CDC, Atlanta, Georgia; Martin Kharrazi, Ph.D., California Department of Health Services, Richmond, California; Muin J. Khoury, M.D., Ph.D., CDC, Atlanta, Georgia; Pamela King, M.P.A., Oklahoma State Department of Health, Oklahoma City, Oklahoma; Katherine Kolor, Ph.D., CDC, Atlanta, Georgia; Rebecca L. Koscik, Ph.D., University of Wisconsin Medical School, Madison, Wisconsin; HuiChuan Lai, Ph.D., University of Wisconsin, Madison, Wisconsin; Larry Lands, M.D., Ph.D., McGill University Health Centre, Montreal, Canada; Mary Lou Lindegren, M.D., CDC, Atlanta, Georgia; Michele A. Lloyd-Puryear, M.D., Ph.D., Maternal and Child Health Bureau, Rockville, Maryland; Thomas H. Locke, M.D., Washington State Board of Health, Olympia, Washington; Jerry McClure, Mississippi State Department of Health, Jackson, Mississippi; Susanna A. McColley, M.D., Children's Memorial Hospital, Chicago, Illinois; Karen McKay, M.D., The Children's Hospital at Westmead, Australia; Catherine McKeon, Ph.D., National Institutes of Health, Bethesda, Maryland; Capt. John A. McQueston, M.D., U.S. Naval Medical Center, Portsmouth, Virginia; Anil Mehta, M.R.C.P., University of Dundee, Scotland; Joanne Mei, Ph.D., CDC, Atlanta, Georgia; Julie Miller, Nebraska Department of Health and Human Services, Lincoln, Nebraska; Micah Milton, M.P.H., CDC, Atlanta, Georgia; Mark Montgomery, M.D., University of Calgary, Alberta, Canada; Cynthia Moore, M.D., Ph.D., CDC, Atlanta, Georgia; Jelili Ojodu, M.P.H., Association of Public Health Laboratories, Washington, DC; Richard Olney, M.D., CDC, Atlanta, Georgia; Richard B. Parad, M.D. Harvard Medical School, Boston, Massachusetts; Kenneth A. Pass, Ph.D., New York State Department of Health, Albany, New York; Suzanne R. Pattee, J.D., Cystic Fibrosis Foundation, Bethesda, Maryland; Rodney J. Pollitt, Ph.D., Sheffield Children's Hospital, Sheffield, United Kingdom; Michael J. Rock, M.D., University of Wisconsin, Madison; Margaret Rosenfeld, M.D., Children's Hospital and Regional Medical Center, Seattle, Washington; Lainie Friedman Ross, M.D., Ph.D., University of Chicago, Illinois; Jack K. Sharp, M.D., State University of New York, Buffalo, New York; Marci Sontag, Ph.D., University of Colorado Health Sciences Center, Denver, Colorado; Brad L. Therrell, Ph.D., National Newborn Screening and Genetics Resource Center, Austin, Texas; Audrey Tluczek, Ph.D., University of Wisconsin, Madison, Wisconsin; Kathy Tomashitis, M.N.S., Columbia, South Carolina Department of Health and Environmental Control, Columbia, South Carolina; Thomas F. Tonniges, M.D., American Academy of Pediatrics, Elk Grove Village, Illinois; Terry B. White, Ph.D., Cystic Fibrosis Foundation, Olympia, Washington; Bridget Wilcken, M.B.Ch.B., The Children's Hospital at Westmead, Australia; Benjamin S. Wilfond, M.D., National Institutes of Health, Bethesda, Maryland; Robert W. Wilmott, M.D., St. Louis University School of Medicine, St. Louis. Missouri.
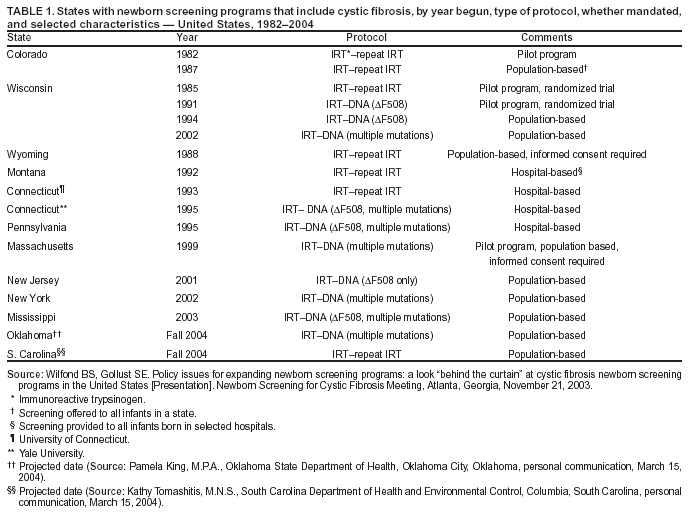
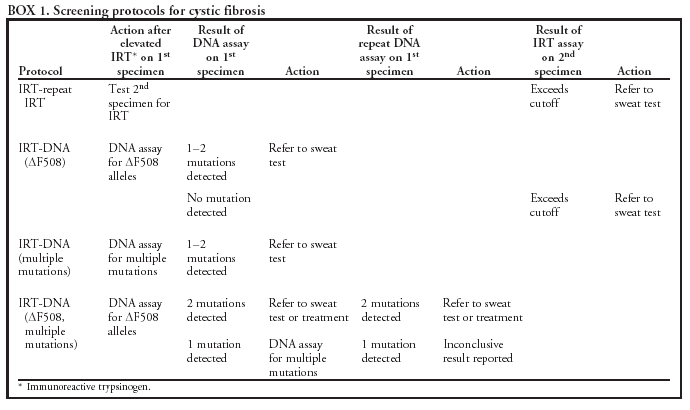
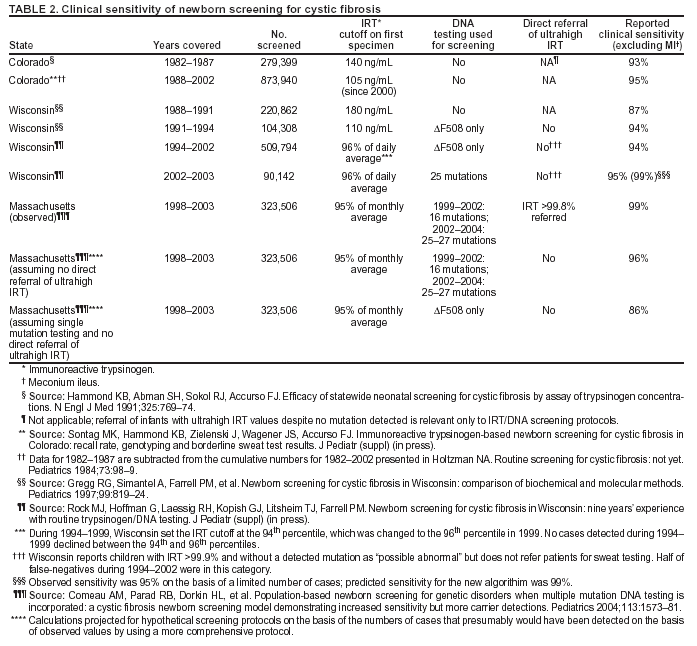
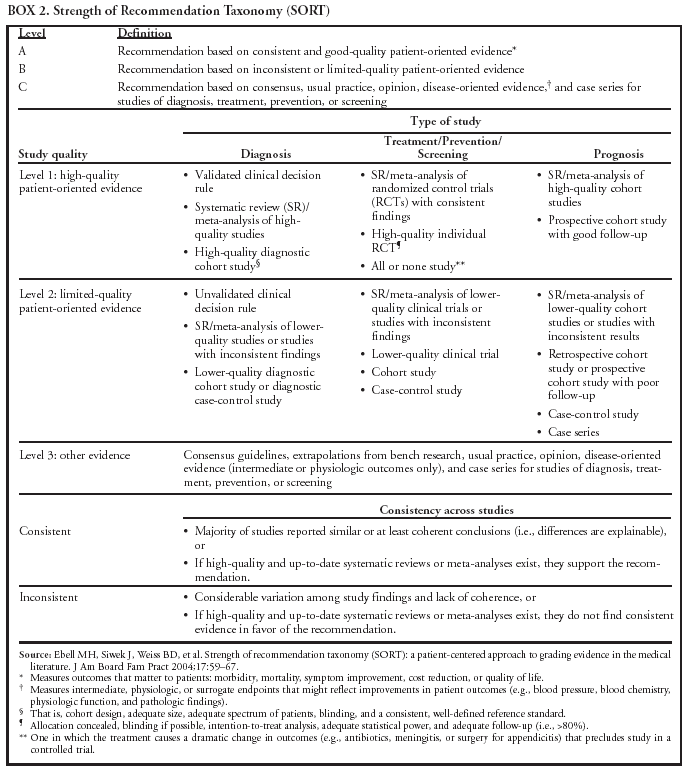
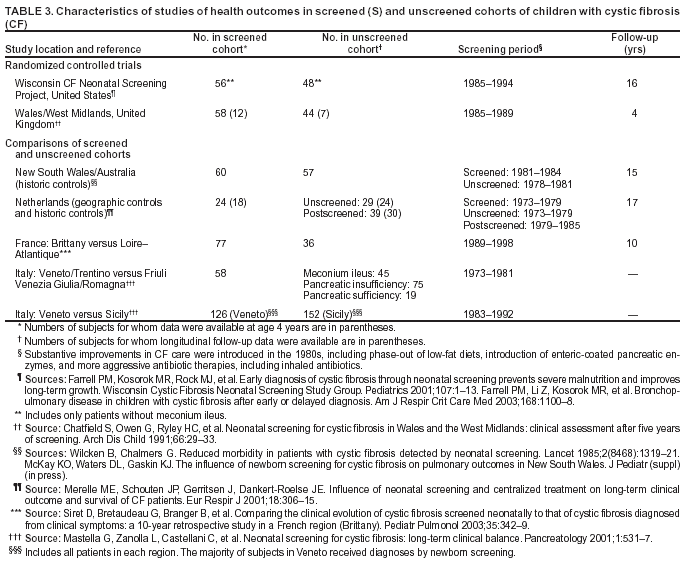
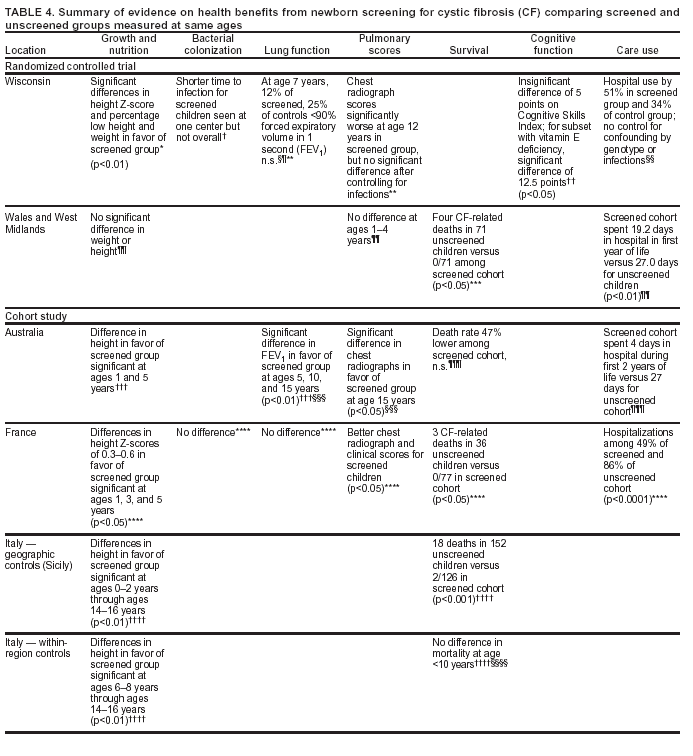
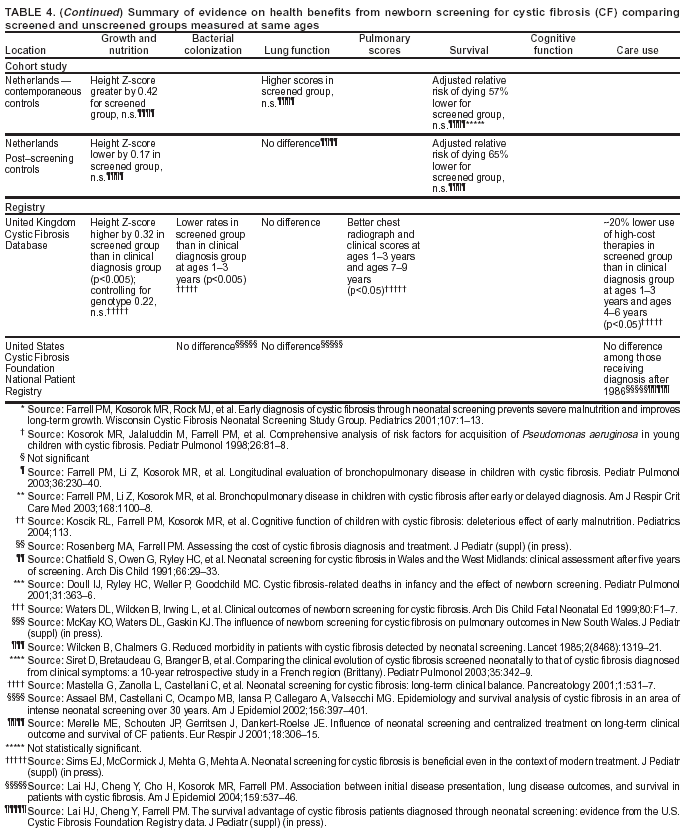
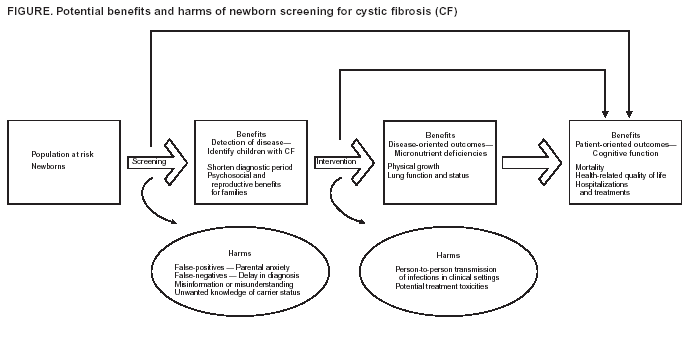
Use of trade names and commercial sources is for identification only and does not imply endorsement by the U.S. Department of
Health and Human Services. |
All MMWR HTML versions of articles are electronic conversions from ASCII text into HTML. This conversion may have resulted in character translation or format errors in the HTML version. Users should not rely on this HTML document, but are referred to the electronic PDF version and/or the original MMWR paper copy for the official text, figures, and tables. An original paper copy of this issue can be obtained from the Superintendent of Documents, U.S. Government Printing Office (GPO), Washington, DC 20402-9371; telephone: (202) 512-1800. Contact GPO for current prices.
**Questions or messages regarding errors in formatting should be addressed to mmwrq@cdc.gov.Page converted: 10/5/2004
|
|
|||||
|
HOME |
ABOUT MMWR |
MMWR SEARCH |
DOWNLOADS |
RSS
|
CONTACT
|
|||||
|
|
|||||
|
This page last reviewed 10/5/2004