Persons using assistive technology might not be able to fully access information in this file. For assistance, please send e-mail to: mmwrq@cdc.gov. Type 508 Accommodation and the title of the report in the subject line of e-mail.
Applying Public Health Strategies
to Primary
Immunodeficiency Diseases
A Potential Approach to Genetic Disorders
Prepared by
Mary Lou Lindegren, M.D.1
Lisa Kobrynski, M.D.2
Sonja A. Rasmussen, M.D.3
Cynthia A. Moore, M.D., Ph.D.3
Scott D. Grosse, Ph.D.4
Marsha Lynne Vanderford, Ph.D.5
Thomas J. Spira, M.D.6
J. Steven McDougal, M.D.6
Robert F. Vogt, Jr., Ph.D.7
W. Harry Hannon, Ph.D.7
Lisa V. Kalman,
Ph.D.7
Bin Chen, Ph.D.8
Marifran Mattson, Ph.D.9
Timothy G. Baker, M.P.H.1
Muin Khoury, M.D.,
Ph.D.1
1Office of Genomics and Disease Prevention,
National Center for Environmental Health, CDC
2Emory University, Atlanta, Georgia
3Division of Birth Defects and Developmental Disabilities,
National Center on Birth Defects and Developmental Disabilities, CDC
4Office of the Director,
National Center on Birth Defects
and Developmental Disabilities, CDC
5Office of the Director, National Center for Environmental Health
6Division of AIDS, STD, and TB Laboratory
Research,
National Center for HIV, STD, and TB Prevention, CDC
7Division of Laboratory Sciences,
National Center for Environmental Health, CDC
8Division of Laboratory Systems,
Public Health Practice Program Office, CDC
9Purdue University, West Lafayette, Indiana
The material in this report originated in the National Center for Environmental Health, Richard J. Jackson, M.D., Director; the Office of Genomics
and Disease Prevention, Muin J. Khoury, M.D., Ph.D, Director; and the Division of Laboratory Sciences, Eric J. Sampson, Ph.D., Director; the
National Center on Birth Defects and Developmental Disabilities, José F. Cordero, M.D., Director, and the Division of Birth Defects and Developmental Disabilities, Gilberto Chavez, M.D., Director; the National Center for HIV, STD, and TB Prevention, Harold W. Jaffe, M.D., Director, and the Division of AIDS, STD, and TB Laboratory Research, Jonathan E. Kaplan, M.D., Director; and the Public Health Practice Program Office, Suzanne Smith, M.D.,
Acting Director, and the Division of Laboratory Systems, Robert Martin, Dr.P.H., Director.
Summary
Primary immunodeficiency (PI) diseases are a group of primarily single-gene disorders of the immune system.
Approximately 100 separate PI diseases have been described, but <20 probably account for >90% of cases. Although diverse, PI diseases share the common feature of susceptibility to infection and result in substantial morbidity and shortened life spans. Most
important, prompt diagnosis and treatment can now lead to life-saving treatment and result in marked improvements in the quality and length of life for persons with PI diseases.
In November 2001, a workshop was convened by CDC in Atlanta, Georgia, to discuss ways to improve health
outcomes among persons with PI disease. A multidisciplinary panel of persons knowledgeable in PI diseases and public health met to identify and discuss public health strategies that can be applied to PI diseases and possibly for other genetic disorders.
A systematic assessment based on the established public health framework was applied to the growing group of PI diseases, whose diverse genetic mutations span multiple components of the immune system but all lead to increased incidence and severity of infections.
During the meeting, specialists in clinical immunology, public health, genetics, pediatrics, health communication, and ethics from state and federal agencies, academic centers, professional organizations, and advocacy foundations discussed the four components of the public health framework as they relate to PI diseases. These four components include 1) public
health assessment (application of traditional public health methods to assess the occurrence and impact of PI diseases on communities); 2) population-based interventions (development, implementation, and evaluation of screening tests administered to newborns and clinical algorithms for early recognition of symptomatic persons to facilitate the earliest possible diagnosis and
treatment for PI diseases); 3) evaluation of screening and diagnostic tools (to ensure their quality and appropriateness for identification of patients with PI diseases); and 4) communication (communication with and information dissemination to
health-care providers and the public to facilitate prompt and appropriate diagnosis and intervention). The working group's
deliberations focused on challenges and opportunities, priority research questions, and recommendations for future action for these four components. These recommendations, developed by workshop participants, will be useful to medical and public
health professionals who are evaluating methods to increase recognition of PI diseases and other genetic
disorders.
Introduction
Advances in human genetics and the evolution of the
Human Genome Project will play a central role in the practice
of medicine and public health in the
21st century. However, gene discovery is only the beginning. For the majority of diseases,
a gap exists between discovering or sequencing genes and using human genomic information to improve health outcomes
(1). Public health research and policy have a crucial role in closing that gap. Moving from gene discovery to clinical and public health application requires full engagement of public health to 1) quantify the effect of genetic discoveries on
population health, 2) develop policies regarding and guidelines for the appropriate use of genetic tests and services, 3) develop
interventions to improve health outcomes, 4) initiate and maintain behavior change among patients and health-care providers, and 5) address the quality of and access to services. Genomic breakthroughs have been identified as
major challenges for public health in the
21st century (2). However, the usefulness of these breakthroughs in clinical practice depends on the availability of population-based data to determine the prevalence of gene variants among different populations,
the population-based risk for disease associated with gene variants, gene-environment interactions, and the effectiveness of genetic tests and services (3--5).
As part of efforts to highlight the emerging role of human genomics in the practice of public health in the United
States, CDC, in collaboration with research, academic, clinical, and foundation partners, evaluated public health strategies that can be used to close the gap between gene discoveries and clinical practice for primary immunodeficiency (PI) diseases
--- approximately 100 primarily single-gene disorders of the immune system. Identification of the genes responsible for these conditions is progressing rapidly; therefore, a
population-based framework is needed that can be applied also to other
genetic disorders and gene discoveries. This report
describes the concerns, challenges, and opportunities and
provides recommendations for public health action regarding such a framework.
Background
With completion of the Human Genome Project, 30,000--35,000 genes have been mapped
(6--9), each of which contains the code for a specific product, typically a protein. Through the proteins they encode, genes determine and regulate all human body processes. Human genomics includes a continuum from the study of single-gene disorders with high penetrance
to common genetic variants or polymorphisms at multiple loci, with low penetrance, and that have complex
gene-environment interactions (10). Genetic disorders are caused by mutations, or alterations, in a gene or set of genes. Mutations can
be inherited or occur de novo. The effect of a mutation on a gene depends on how it alters the expression or function of the
gene product and the role of that protein in the body. Mutations in certain genes have severe effects, whereas mutations in others
do not.
The majority of genetic disorders result from a complex interplay of multiple genetic changes and environmental
factors. However, certain disorders result when a mutation alters or causes an absence of the product of only one gene.
Examples of such single-gene disorders are cystic fibrosis (CF) and phenylketonuria (PKU). Single-gene disorders can be
either X-linked (i.e., caused by a defect in a gene on the X chromosome) or autosomal (i.e., caused by a defect in a gene on an autosome or nonsex chromosome). Single-gene disorders can result from either dominant or recessive patterns of inheritance or
expression. Selected chromosomal disorders, which might be inherited, involve microdeletions of multiple genes at closely linked
loci. Although single-gene disorders are individually rare, they collectively contribute to a substantial proportion of pediatric morbidity and mortality (1).
PI diseases are a group of primarily single-gene disorders of the immune system
(11--13). Primary denotes the
genetic nature of the defects, differentiating them from
secondary, or acquired, immunodeficiencies caused by malnutrition,
infection (e.g., human immunodeficiency virus [HIV] infection), chemotherapy, or other external agents. Approximately 100
separate PI diseases have been described, but <20 probably account for >90% of cases. The disorders vary in the severity and spectrum of symptoms, but without effective and early treatment, they can be fatal. A high index of suspicion and prompt diagnosis can lead to lifesaving treatment and substantial improvement in quality of life for persons with PI diseases. Causes of PI diseases vary, but single-gene defects can lead to a missing enzyme, a missing structural component, developmental arrest at a
specific differential stage of immune development, or a nonfunctional protein. As with all single-gene disorders, selected PI diseases are known to be X-linked or autosomal, with both dominant and recessive patterns of inheritance or de novo mutations;
others might have more complex modes of inheritance not yet understood. Approximately 80% of affected persons are aged
<20 years, and because certain PI diseases are inherited in X-linked
recessive fashion, 70% of cases occur among males
(13).
Advances in human genomics have led to identification of the gene defects responsible for >60 PI diseases and
have prompted development of new diagnostic and therapeutic tools and potential gene therapies
(14--20). New molecular techniques have facilitated identification of different types of mutations underlying PI diseases. Single-nucleotide substitutions, or point mutations, involve an alteration in the sequence of nucleotides in a gene. These include missense mutations, which alter the amino acids in the protein product of a gene; nonsense mutations, which generate premature stop codons in the genetic code; RNA (ribonucleic acid) splice-site mutations, which can lead to frameshift mutations;
and
regulatory mutations, which affect aspects of gene expression. Mutations also can involve insertions or deletions of
DNA (deoxyribonucleic acid) sequences. Progress in the delineation of the mechanisms by which these genetic mutations cause
PI diseases has added to the understanding of the normal immune system and the processes that underlie conditions that
occur with far greater frequency than PI disease
(21).
Clinical Characteristics and Effect of PI Diseases
The clinical hallmark of PI diseases is an increased susceptibility to infection, the severity of which varies by defect
(13,22). In certain cases, the body fails to produce any or sufficient antibodies to fight infection. In other cases, the cellular (e.g., T-cell) defenses against infection fail to work properly. Shared features of the disorders are an unusual rate or severity of
infection, infection with unusual or opportunistic organisms, and infection associated with specific syndromes
(13). PI diseases also are associated with other immunologic disorders (e.g., autoimmune diseases) and carry an increased risk for cancer, particularly lymphoid malignancies (22). PI diseases often are classified according to the affected components of the immune
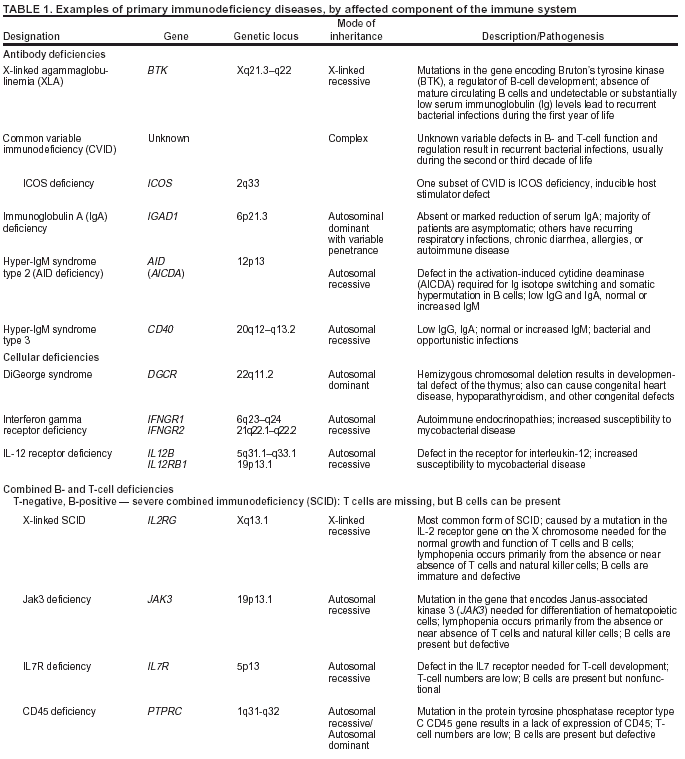
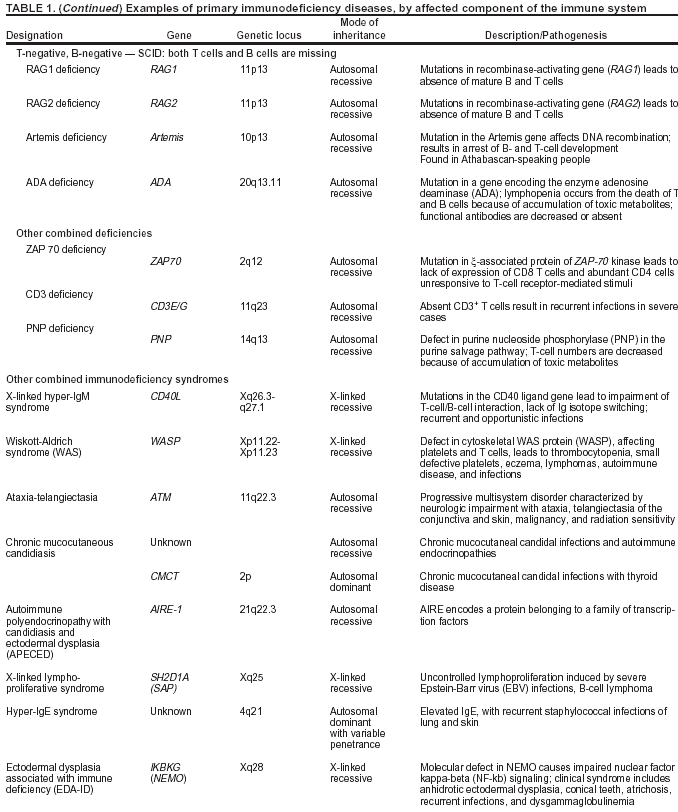
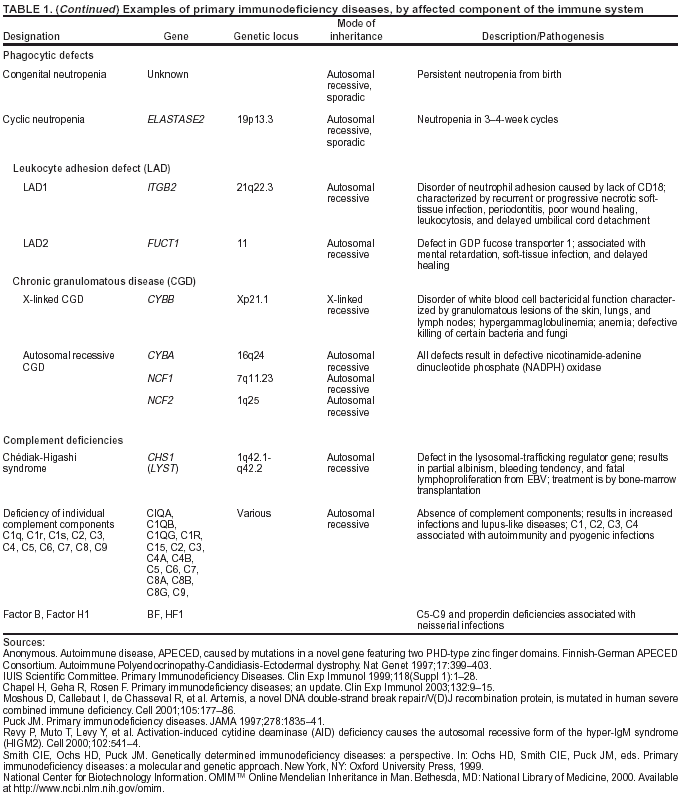
system (Table 1).
Antibody Deficiencies
Approximately half of the diseases are associated with inadequate or defective antibody production, caused by too
few antibody-producing B cells or B cells that do not function properly, resulting in inadequate production of
antigen-specific antibodies (23). These disorders are characterized by recurrent sinus and pulmonary infections and septicemias with bacteria (13,24). The most severe defect in this category is
X-linked agammaglobulinemia (XLA), typified by a
limited number or no mature B cells or antibody-secreting plasma cells. Affected persons develop severe, recurrent bacterial infections, usually during the first year of life.
Other antibody defects are common variable immunodeficiency (CVID) and immunoglobulin A (IgA) deficiency. CVID
is characterized by variably low levels of immunoglobulin G (IgG), immunoglobulin M (IgM), and IgA, and
suboptimal antibody responses after vaccination. CVID patients usually experience recurrent bouts of pneumonia and infections of
the joints, bones, and skin. These persistent infections lead to organ damage, often resulting in disability or death from
chronic lung disease (25). Moreover, affected
females with CVID had a >400-fold increased risk for lymphomas in their fourth
and fifth decades of life compared with age-matched general population risks in one U.S. study
(25). IgA deficiency, similar to other PI diseases, has a wide clinical spectrum. Although all affected persons lack IgA in the
mucous membranes lining the airways and digestive tract, certain persons are asymptomatic whereas other have recurrent infections. For reasons not completely understood, the incidence of allergy or autoimmune disease is increased among patients with selective
IgA deficiency. Certain IgA-deficient persons might have severe or fatal anaphylactic
reactions to blood or blood-products containing IgA.
Combined B- and T-Cell Deficiencies
Combined B-cell and T-cell immunodeficiencies constitute approximately 20% of PI diseases
(23). In the most serious forms (e.g., severe combined immunodeficiency [SCID] disorders), survival beyond the first year of life is rare without prompt immune reconstitution through hematopoietic stem cell transplantation
(15,16,19,26,27). Immune reconstitution with
gene therapy has been achieved for forms of SCID
(14,20). Early diagnosis of SCID is critical because the chances for
successful treatment are highest for infants who have not yet experienced severe opportunistic infections (19). Mutations in eight different genes cause SCID
(19,28). Approximately half of all cases are linked to the X chromosome. X-linked SCID
results from a mutation in the interleukin 2 receptor gamma
(IL2RG) gene that produces the common gamma chain subunit, a component of multiple IL receptors. The product of the IL2RG gene activates a key signaling molecule, Janus-associated kinase 3 (JAK3 gene product). A
mutation in JAK3 also can result in SCID. Other forms of SCID are associated
with deficient activity in the enzyme adenosine deaminase
(ADA gene product) or a defect in the recombination-activating
gene (RAG). The genetic defect has not been identified for certain forms of SCID. Other combined immunodeficiencies are part of well-defined immunodeficiency syndromes (e.g., Wiskott-Aldrich syndrome [WAS], ataxia telangiectasia, and
hyper-IgE syndrome), all of which are associated with recurrent infections and decreased life
expectancy (Table 1).
Cellular immune deficiencies, resulting from defects in
T-cell maturation or function, contribute an estimated 10% of
PI cases (23). One example is DiGeorge syndrome, which is typified by aberrant development of the heart, parathyroid glands, or
thymus. The absence of a thymus gland in patients with DiGeorge syndrome leads to low T-cell numbers and
decreased function, but the degree of immunologic impairment varies considerably
(29,30). Approximately 90% of these patients have
a microdeletion in chromosome 22q11.2, such that multiple genes from this region are absent (additional information
is available at http://www.genetests.org).
Defective Phagocytes
An estimated 18% of PI cases result from defective phagocytes
(23). Phagocytic defects result in the inability of cells
that normally engulf and kill invaders to remove pathogens or infected cells from the body. Chronic granulomatous
disease (CGD), caused by a defect in intracellular killing of bacteria by phagocytes, usually appears in childhood, but milder forms can appear in the second or third decade of life. It can be inherited as an X-linked or autosomal-recessive defect; affected persons experience frequent and severe infections of the skin, lungs, and bones and tumor-like masses called granulomas. In leukocyte adhesion defect (LAD), phagocytes lack an
essential adhesion molecule, preventing them from migrating to sites
of infection. The result is recurrent, life-threatening infections, especially of the soft tissues. Chédiak-Higashi syndrome is a rare and usually fatal disorder caused by granule defects in phagocytes, platelets, and melanocytes. Patients have
partial oculocutaneous albinism and often experience overwhelming and fatal infections with Epstein-Barr virus. Both LAD
and Chédiak-Higashi syndrome are inherited as
autosomal-recessive defects.
Complement System Defects
Defects in the complement system occur less frequently than other PI diseases. They are associated with a
nonfunctional protein or the absence of a complete complement molecule capable of attaching to antibody-coated foreign invaders and opsonizing bacteria. The most common defect, C2 deficiency, is an autosomal-recessive inherited defect in the gene for
the complement protein C2. Affected persons have recurrent and severe infections with encapsulated bacteria,
frequently meningitis, and a susceptibility to autoimmune diseases. Terminal complement protein (C6-8) deficiencies are associated with severe infections with Neisseria
meningitidis and N. gonorrhoeae.
Prognoses for Patients with PI Diseases
Although PI diseases share selected clinical manifestations, both the timing of the onset of symptoms and the prognosis vary considerably. Patients with antibody or complement
deficiencies can have near-normal life spans, if their deficiencies are diagnosed early, managed appropriately, and are not
affected by concurrent chronic diseases. Persons with
phagocytic disorders, combined immunodeficiency disorders, and antibody disorders with chronic infections have guarded prognoses; the majority are chronically ill and require intensive treatment. Certain severe PI diseases (e.g., SCID) become apparent early in life, with only a short asymptomatic period after birth. Without an effective early intervention, the
majority result in death during the first years of life.
Incidence and Birth Prevalence Estimates
The true frequency of PI diseases in the general population, either individually or in the aggregate, has not been
ascertained, but estimates have been reported. Certain countries have developed registries to collect information regarding cases of PI diseases (31--36). The minimum prevalence of PI has been estimated by using data collected from these registries. At least five factors cause these registries to underestimate the true prevalence of PI diseases: 1) lack of clinical recognition, 2) lack
of reporting to the registries, 3) overrepresentation of certain referral centers, 4) lack of a standardized case definition, and 5) death before recognition. Population-based data related to incidence and prevalence are critically needed.
The reported minimal estimate of birth prevalence of SCID based on recognized cases is 1/100,000, but this under-estimates the prevalence because of infant deaths occurring before diagnosis
(15). In contrast, selective IgA deficiency, the
most common immunodeficiency, was found in as many as 1/328 healthy blood donors
(37). In aggregate, the estimated incidence of diagnosed PI diseases has been reported as
1/10,000 persons (22,38,39). As a comparison, incidence
estimates for CF are 1/2,500 among whites and for PKU are 1/16,000 persons
(40,41).
Diagnosis
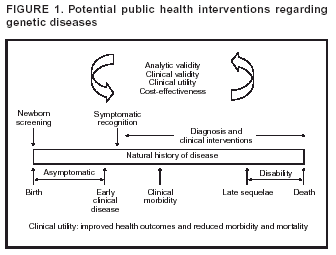
Early detection is possible for the majority of PI diseases, is critical for the success of certain therapies, and can be life-saving. Genetic diseases (e.g., single-gene disorders with high penetrance) can be detected along a continuum of
symptomatic expression by using 1) screening tests to evaluate asymptomatic newborns for conditions that require early intervention and 2) clinical algorithms for early recognition of symptomatic persons before the onset of clinical morbidity, with
confirmatory laboratory diagnosis (including genetic testing) (Figure 1). Effective treatment regimens then can be initiated early in the course of disease to reduce morbidity, disability, and mortality.
The first clinical clue in diagnosis of a PI disease is usually a history of infections that are persistent, recurrent, difficult
to treat, or caused by unusual microbes. Because PIs are frequently inherited, a positive family history is also a key diagnostic tool (42); in a series of 70 PI patients identified in an immunology clinic, 18.6% (N = 13) had family histories
of immunodeficiency (43). The type of infection identified in either the
patient or the family history also might indicate
the nature of an immunodeficiency. Infections with bacterial organisms are frequently observed among patients with
antibody deficiencies; severe infections from viruses, fungi, and other opportunistic organisms characterize T-cell immunodeficiencies. Recurrent infections with staphylococcal and other catalase-positive organisms indicate phagocytic defects, and recurrent Streptococcus pneumoniae or
Neisseria infections characterize patients with complement deficiencies.
Physical examination can identify characteristic physical findings and anatomic changes secondary to
infections. Patients with PI diseases often appear chronically ill, with pallor, malaise, and a distended abdomen caused
by hepatosplenomegaly. Patients with XLA typically lack peripheral lymph nodes, adenoids, and tonsils. Lymphadenopathy is observed frequently among patients with CGD. In WAS, the genetic mutation causes thrombocytopenia as well as
immune defects; children have bruising, petechiae, and eczematous rash
(44). However, clinical symptoms can vary from patient
to patient, even for identical mutations of the same gene
(45). Typical radiographic findings include an absent thymus, which
is the hallmark of DiGeorge syndrome and multiple types of SCID. Children with infant-onset ADA deficiency often have characteristic skeletal abnormalities of the ribs and hips readily apparent on radiograph.
Laboratory tests are required to diagnose a PI disease
(46). No single testing modality is appropriate for all situations.
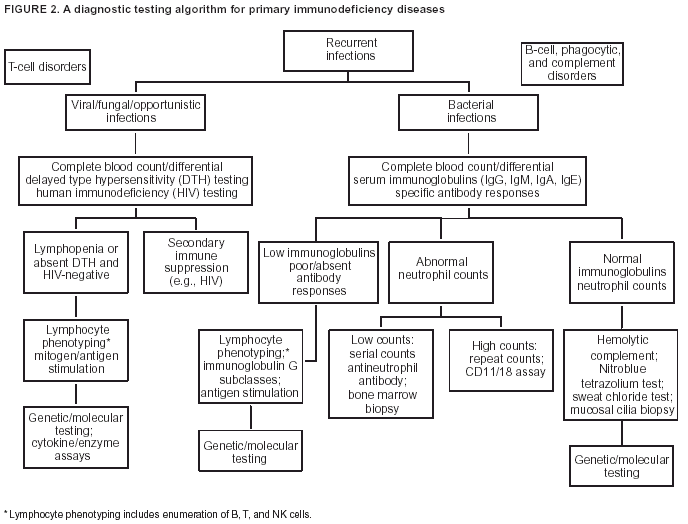
Given that certain PI diseases have overlapping features and that selected ones can be caused by combined immune defects, clinicians advocate a stepwise approach to screening the
immune system (Figure 2). The majority of initial tests are available
through commercial or hospital laboratories and include
tests to assess humoral immunity (i.e., Ig proteins and specific
antibodies), cellular immunity (e.g., lymphocyte/mononuclear cell quantitation or functional assays), phagocytic cell function,
and complement components and function.
Genetic testing involves "analysis of human DNA, RNA, chromosomes, proteins, and certain metabolites to detect
heritable disease-related genotypes, mutations, phenotypes or karyotypes for clinical purposes"
(47). In cases for which the location of the genetic defect is known, testing involves direct testing of the patient's DNA to identify specific mutations. In certain cases, an assay to measure mRNA (messenger RNA) (e.g., polymerase chain reaction [PCR]) or the protein product
(e.g., immunoblotting or flow cytometry) can confirm a diagnosis when the gene product is absent; however, this method
cannot detect disease associated with a nonfunctional protein. A simple, reliable way to evaluate function for T cells is delayed
type hypersensitivity skin tests and for B cells, antibody responses
after vaccination.
Treatment
Interventions for PI diseases are aimed at preventing infection, prolonging life, and improving quality of life
(48). Use of antibiotics to treat and prevent infections is a key element in patient management. In certain cases, prophylactic
antibiotics help to prevent infections (e.g., trimethoprim-sulfamethoxazole to prevent
Pneumocystis carinii pneumonia among
patients with T-cell defects and prevent recurrent
infections among patients with CGD). Research has demonstrated the safety
and efficacy of replacement therapy with intravenous Ig (IVIG) among patients with defects in antibody production (49). Enzyme replacement therapy for ADA deficiency also is effective
(50). Curative interventions, primarily bone-marrow and
stem-cell transplantation, have been used with varying degrees of success for an expanding array of PI diseases (15,16,19,26,27,51,52). Clinical trials also have demonstrated that gene therapy can restore near-normal immune function among patients with SCID caused by
mutations in IL2RG, and similar types of therapy are promising for other immunodeficiencies
(14,17,20). However, recently, the occurrence of T-cell leukemia in two of 10 children administered gene therapy for
IL2RG SCID (mutant gamma-
chain IL-2 receptor) has prompted a halt to all gene therapy using retroviral vectors for immunodeficiency. In these cases,
the retroviral gene construct of the IL2RG gene inserted itself on the oncogene
LMO2 that is aberrantly expressed in acute lymphocytic leukemia of childhood. Thus, insertional oncogenesis was the probable cause of the T-cell leukemia in these two cases (53--55).
Public Health Framework
The defining characteristics of PI diseases make them candidates for a public health intervention approach. Although
the clinical manifestations and underlying genetic defects are diverse, PI diseases share the common feature of
increased susceptibility to infection and collectively result in substantial morbidity and shortened life spans. Most important,
prompt diagnosis and treatment can be life-saving and result in marked improvements in the quality and length of life.
The foundation for a public health intervention to improve the health status of persons with PI diseases is
population-based information regarding the incidence, prevalence, and natural history of the diseases; the accuracy of diagnostic methods;
and the efficacy of early interventions. However, the majority of these data are lacking. The heterogeneity of PI diseases and the limited understanding of the relation between genotype and phenotype also hinder intervention efforts. Additional
obstacles include the difficulty of diagnosis in the
absence of a high index of suspicion and the lack of awareness among
health-care providers and the public, which impedes the timely recognition of affected persons by using a combination of clinical suspicion and diagnostic testing.
To address these impediments and improve health outcomes among persons with PI diseases, CDC and partners
have adapted a population-based public health framework developed as part of CDC's strategic plan for genomics and
public health, to the problem of PI diseases
(56). The framework has four components as follows:
public health assessment --- application of traditional public health methods to assess the impact of PI diseases on community health;
population-based interventions --- development, implementation, and evaluation of screening tests administered
to newborns and clinical algorithms for early recognition of symptomatic persons to facilitate the earliest possible
diagnosis and treatment for PI diseases;
evaluation of screening and diagnostic tools --- evaluation of screening and diagnostic tools to ensure their quality and appropriateness for identification of patients with PI diseases; and
communication --- communication with health-care
providers and the public to facilitate prompt and appropriate
diagnosis and intervention.
CDC has begun to apply this framework in the context of ethical, legal, and social considerations to different
conditions, most recently to hereditary hemachromatosis, a treatable, adult-onset, single-gene disorder of iron metabolism (57--61). For example, gaps in data related to the natural history of the disease, penetrance, optimal treatment for asymptomatic persons, and the psychosocial effect of genetic testing precluded recommendations for population screening for mutations in
HFE, the associated gene (62--64). However, educational efforts are under way to facilitate early diagnosis (e.g., iron overload and
HFE mutation testing). Lessons learned from applying the framework to hemochromatosis is being applied to other
conditions, including PI diseases.
In November 2001, CDC convened a multidisciplinary panel of specialists to identify and discuss public health
strategies that can be applied to PI diseases and also used as an approach for other genetic disorders
(65). A systematic assessment based on the established public health framework was applied to the growing group of recognized PI diseases, for which diverse genetic mutations span multiple components of the immune system but all lead to the increased incidence and severity
of infections. During the meeting, specialists in clinical immunology, public health, genetics, pediatrics, health
communication, and ethics from state and federal agencies, academic centers, professional organizations, and advocacy foundations
discussed the public health framework as it relates
to PI diseases. The working group's deliberations were organized around the
four components of the framework and centered on challenges and opportunities, priority research questions,
and recommendations for public health action. The remainder of this report reflects their analysis of the problem,
their conclusions and recommendations, and subsequent
deliberations and findings.
Public Health Assessment
Assessment Tools
The majority of what is understood regarding PI diseases derives from accumulation of data from clinical case reports,
case series, and case registries. This approach has advantages but has not provided a complete understanding of the
incidence, prevalence, and natural history of PI diseases. A public health assessment of the magnitude and characteristics of the problem in the United States, using population-based data, is needed. Quantitative public health methods can be used to assess the effect of gene variants on the risk for disease, disability, and death and to determine the impact of
population-based interventions on improved health outcomes. The traditional tools of public health assessment are 1) surveillance, 2) epidemiology, and 3) laboratory science.
Surveillance Systems. Surveillance is the systematic collection, analysis, and interpretation of data related to
health outcomes and other health-care events for use in planning, implementation, and evaluation of population-based
health activities (66,67). Surveillance data can be derived from traditional data sets (e.g., vital records and health surveys) or obtained proactively from health-care providers, health-care institutions with electronic patient records, or laboratories.
Effective surveillance requires standardized case definitions for each disorder of interest.
A surveillance system for PI diseases should be used to
determine the incidence and prevalence of these
conditions. Assuming routine performance of genotyping, a laboratory-based surveillance component should facilitate the calculation
of the prevalence of gene variants among cases. The ability to link cases with other data sets will help determine the
morbidity, mortality, disability, and health-care costs associated with PI diseases and help set priorities based on public health impact. The availability of outcomes data will allow evaluation of the effect of changes in health-care policy and
practice.
Epidemiologic Research. Epidemiology is the study of the distribution and determinants of disease in
specified populations, including assessment of the causal effect of preventive interventions on health outcomes. Although
clinical research can identify gene variants and other risk factors for PI diseases, population-based analytic epidemiologic studies
are needed to quantify the effect of gene variants on the risk for disease, death, and disability and to determine the
relations between genotype and phenotype in the population
(1). Epidemiologic studies that contribute to the understanding of
the natural history and clinical course of PI diseases and the benefits of early detection and intervention can improve
individual outcomes and reduce the public health burden of this group of diseases. Epidemiologic research methods also are needed to assess the determinants and uses of genetic testing and other promising interventions and health-care practices.
Laboratory Science. Both surveillance and epidemiologic research are conducted in conjunction with laboratory
efforts. These center on diagnostics, phenotypic characterization, genetic analysis, studies of genotype-phenotype relations, and development and evaluation of screening and diagnostic tests.
Existing Data-Collection Systems
Existing population-based data from which to derive a public health assessment of PI diseases are limited. Available data are derived from case-based disease registries that collect patient-specific information from multiple sources.
Disease and Mutation Registries. Case-based registries usually are designed to improve patient care but can be helpful
for studying rare diseases. In 1992, the Immune Deficiency Foundation (IDF) initiated a registry of U.S. patients with CGD and 5 years later expanded the project to include seven other disorders --- hyper-IgM syndrome, XLA, CVID, WAS, SCID,
LAD, and DiGeorge syndrome (36). The most reliable data from these registries are for CGD, for which IDF has calculated
a minimum estimated U.S. incidence of 1/200,000 live-born infants
(36). The registry also is used to collect data related
to natural history and clinical course, including the response to treatment. In 1995, IDF conducted a national,
cross-sectional survey of approximately 17,000 immunologists
and medical school faculty to estimate the burden of PI diseases in the
United States, to describe characteristics of persons with these disorders, and to identify problems related to access to
treatment. Approximately 1,500 physicians reported caring for an estimated 21,000 patients with PI disease
(68).
Other countries have developed their own registry-based estimates of the frequency of PI diseases, ranging from
an estimated prevalence of 2.1/100,000 in Australia
(31) to 6.8/100,000 in Norway
(32--34). A registry maintained by the European Society for Immunodeficiencies (ESID) collects data regarding patients from approximately 25 countries in Europe (69). As of July 2000, the ESID registry contained clinical data for approximately 8,900 patients from 26
countries
(70). An example of a registry for another genetic disorder that might be a model for PI diseases is the CF registry, which
is based on case ascertainment at comprehensive treatment
centers. The Cystic Fibrosis Foundation (CFF) sponsors the National Cystic Fibrosis Patient Registry to collect data
regarding all patients examined at CFF-supported and accredited care centers (71). Data are used to support epidemiologic studies, direct research, and design clinical trials, all with the goal of improving the survival of persons with CF
(72).
Other sources of case-based information are the Internet-based, locus-specific immunodeficiency mutation databases established by ESID and expanded by other investigators
(73,74). These databases contain information regarding
specific mutations and certain clinical features of affected persons. The first Internet-based immunodeficiency mutation
database, BTKbase, was initiated in 1995 to collect information related to mutations in the
BTK gene (Bruton's tyrosine kinase), which causes XLA
(75). Similar locus-specific mutation databases have been developed since then
(69,73). Mutation databases can be used to analyze the types of mutations and their distribution in exons and introns, including their location in protein domains. Mutation databases that contain clinical information can be helpful in assessing
genotype-phenotype relations and determining the presence of gene
variants in asymptomatic family members (76).
Data from disease and mutation registries can be used to estimate the minimal incidence of a disorder, characterize epidemiologic features, and define a range of clinical characteristics in a cohort of patients (36). However, although each has its applications, current registries provide incomplete population-based data regarding the burden of PI diseases.
Continued growth of disease and mutation registries relies on the submission of case reports by physicians, resulting in overrepresentation of certain clinical centers in the sample collection
(59). Incomplete ascertainment limits the representativeness of the
data. Moreover, the lack of standardized case definitions precludes the calculation of sound
population-based rates from these sources. The value of mutation databases for public health assessment also is limited by the rarity of genetic laboratory confirmation of PI diagnoses. In other cases, the mutated sequence might be known but not submitted to the database.
Population-Based Morbidity and Mortality
Data. To contribute to the study of the impact of single-gene disorders,
existing population-based data sources were reviewed. Surveillance databases already have been used to evaluate the
impact of hereditary hemochromatosis (59). Hospital discharge data provide information concerning short-stay hospitalizations
for specific conditions and have been used, for example, to document the substantial morbidity rate and hospitalization
charges associated with birth defects and genetic diseases among children
(57,77,78). However, the national hospital discharge
survey enumerates hospital discharges rather than individual patients, and for rare or underdiagnosed diseases might provide
more limited information because of potential inaccuracy of coding and duplication caused by multiple hospitalizations for the same patient. Managed care organizations maintain substantial, linked, computerized inpatient and
outpatient databases that can be helpful in determining incidence rates
(79). One example is the Vaccine Safety Datalink (VSD), a partnership
between CDC and four health-maintenance organizations designed to evaluate vaccine safety among children. Computerized data concerning vaccinations, medical outcomes, and health services usage are provided for a well-defined population of approximately 1 million children (1993--1996). In addition to determining
vaccine-related adverse events, this database
could be examined for other relatively infrequent events, including PI diseases
(78--80).
Mortality data can provide population-based information concerning survival and cause-specific mortality regarding
genetic disorders (60,81--83). Since 1968, CDC's National Center for Health Statistics has compiled data from all death certificates filed in the United States and made these data available in Multiple-Cause Mortality Files
(82,84). The files include demographic and geographic information regarding the decedent and International Classification of Disease
(ICD) codes for the underlying cause of death and <20 conditions listed on the death certificate
(85,86). Methodologic limitations include reliance on coding systems that are not unique or specific enough for birth defects and genetic diseases; delay between death and availability of data; and limited information regarding risk factors. Despite these limitations, mortality files
and other population-based data sources will be critical for planning interventions for PI diseases, especially as the causes and treatments of these disorders are further elucidated by epidemiologic studies and human genome research (75,87,88).
Population-Based Disease Surveillance. Efforts to collect population-based epidemiologic and surveillance data related
to patients with other genetic diseases also might be helpful models for assessment of PI diseases. Population-based birth-defects surveillance systems also hold promise for collection of data regarding PI diseases
(87). Each state has a different approach
to birth-defects surveillance. Data sources include vital records, hospital and clinic records, and administrative databases. The diversity of approaches --- particularly methodologies used to generate timely data, applications to monitor
prevention
activities, and projects to improve access to health services and early intervention --- provides useful
resources for developing surveillance systems for other childhood diseases.
CDC's program to prevent complications from hemophilia and other bleeding and clotting disorders includes a
national surveillance system, prevention interventions conducted through a nationwide network of hemophilia treatment centers (HTCs), and epidemiologic and prevention research. CDC's first state-based surveillance effort was designed to
identify all patients with hemophilia in six states, characterize the patient population, and identify risk factors and outcomes of care (89,90). Through this effort, CDC derived the first
population-based estimate of hemophilia prevalence in the United
States and demonstrated the effectiveness of the HTC model. In 1996, to address gaps in this system (e.g., lack of patient follow-up and specimen collection), CDC and the HTCs initiated a prospective universal data collection (UDC) system. The UDC system is designed to guide clinical practice, monitor blood safety, develop a specimen repository, and monitor the clinical extent and progression of joint disease
(91). Although the UDC system is more comprehensive than the initial
surveillance effort, the requirement for informed consent might affect its population-based representativeness.
Workshop Recommendations for Action. The goal of public health assessment for PI diseases is to collect population-based data to define the incidence and prevalence of the disorders. Recommendations from the workshop for public health assessment for PI diseases include the following:
Collect population-based data regarding the incidence, prevalence, and natural history of PI diseases.
Collect population-based data regarding the relations
between genotype and phenotype for these diseases.
Collect population-based data regarding the effect of early recognition and effective therapies on morbidity and mortality.
Target three subsets of PI diseases as priorities for a
systematic public health assessment; possibilities include
--- profound T-cell defects, because of their resulting high mortality
in the absence of interventions;
--- antibody deficiencies, because of the substantial
number of persons affected and the high burden of morbidity; and
--- CGD, because of the existence of an established IDF data set.
Conduct pilot activities to improve the collection, use, and quality of surveillance and epidemiologic data. These might include
--- convening a working group of clinical immunologists and scientists to provide guidance regarding case definitions
for registry and surveillance activities;
--- developing collaborations between public and private advocacy groups to expand data collection and completeness
of disease registries and to conduct further analyses;
--- exploring use of existing population-based databases for their potential in yielding useful information
regarding the incidence, prevalence, and natural history
of PI diseases; and
--- developing collaborative state-based surveillance
activities for genetic diseases, including PI diseases. For the short
term, these might include implementing pilot surveillance systems, similar to birth-defects surveillance, in states with large population sizes because of the estimated rare incidence of these diseases. In addition, linking surveillance to existing databases should be explored (e.g., Vaccine Safety Datalink, hospital-discharge data, IDF registry, or laboratory-based reporting). In the future, surveillance can be expanded beyond the pilot states.
Participate in ICD revisions to promote development of unique and specific codes for PI diseases.
Promote development of a network of centers of excellence, and encourage the use of these centers for epidemiologic data collection, specimen repository, and special studies. Possibilities for special studies are longitudinal
spectrum-of-disease studies, clinical trials, and evaluations of genotype/phenotype relations.
Population-Based Interventions
Two major areas were discussed at the workshop, 1) early clinical recognition of PI diseases and 2) newborn screening.
Early Clinical Recognition
Background and Rationale. Timely and effective
population-based interventions can reduce morbidity and mortality
from genetic diseases (Figure 1). For PI diseases, these interventions center on early diagnosis and implementation of
effective therapy (e.g., hematopoietic stem cell transplantation, Ig
replacement, and administration of antibiotics). The
intervention component of the public health framework for PI diseases therefore involves development of strategies for early diagnosis,
implementation of pilot demonstration projects, and evaluation of the effect of these interventions on morbidity,
disability, health-care costs, and mortality.
When evidence indicates that early diagnosis and treatment will avert the late stages of disease and prevent
morbidity, disability, and premature mortality, increased early clinical recognition is one component of a public health response. The goal is to identify persons who have early symptoms indicative of a PI disease so they can receive diagnostic testing to confirm
the presence or absence of disease and receive appropriate interventions to prevent adverse outcomes. Although data regarding
the benefits of early symptomatic screening are limited, information from clinical centers supports improved outcomes for
certain PI diseases through early intervention
(25,92--94). The effect might vary, depending on the genetic defect, the age
at diagnosis, presence of prior infections, and history of vaccination and blood transfusion
(93).
Symptom-Based Screening --- Clinical
Algorithm. Increasing early symptomatic screening for PI
diseases requires concerted efforts to increase awareness of these conditions among physicians and health-care systems.
Primary-care clinicians, particularly pediatricians and family practice physicians, provide the first point of contact for persons with PI diseases by recognizing the possibility of an immunologic problem and the need for appropriate evaluation. Clinicians need
to be aware of the estimated prevalence of PI diseases, the natural history of the disorders, the availability and efficacy of treatment, and most importantly, the common early symptoms. Early recognition of PI diseases in the clinical setting can be facilitated by development and evaluation of a symptom-based screening algorithm. Such an algorithm can be designed to 1) identify persons with a frequency of infections who fall outside the normal range of infections;
2) increase physicians' awareness of the types, frequency, and appearances of PI diseases; 3) facilitate physicians' understanding of useful screening approaches (e.g., family history); and 4) trigger
appropriate action without overburdening the medical
care system.
The enhanced early clinical recognition approach has multiple advantages. Symptom-based screening occurs in the
usual health-care setting and requires no additional screening infrastructure. Although certain children and adults seen in primary-care settings might have clinical symptoms suggesting PI disease, the number tested still will be considerably lower than that required for universal screening. Finally, including
a PI disease as a suspected diagnosis will occur in a clinical
setting that offers options for follow-up and referral.
However, the benefits of the symptom-based approach will be limited if diagnostic testing and treatment are unavailable
or delayed. For example, researchers at Mt. Sinai School of Medicine are studying whether PI diseases are
underrecognized among minority and economically disadvantaged persons. The percentage of white non-Hispanic patients among whom PI diseases are diagnosed and treated at Mt. Sinai is disproportionately high (92%), compared with the population of
the hospital's catchment area of East Harlem, which is predominately Hispanic (52%) and black non-Hispanic (37%).
Possible reasons for the disparity include receipt of care in emergency departments and clinics with multiple providers, lack of regular contact with a primary-care physician, and lack of continuity of care. Investigators are evaluating use of profiles of diagnostic codes that might indicate probable PI diseases and help providers identify patients earlier. Improvements in the specificity and accuracy of coding have been identified as needs. Mt. Sinai also is undertaking outreach and educational efforts
directed toward providers serving minority populations to increase their awareness and improve the timely diagnosis of PI diseases (65). Such efforts at other centers and in a population-based approach might substantially affect the care of patients with
PI diseases.
Assessment and Evaluation of Impact. Initiation of treatment after identification of a PI disease and early in the course
of disease might be sufficient to prevent premature mortality, but a patient's quality of life will not improve if the sequelae are not reversible or the disease progression cannot be halted. Thus, systematic studies of the natural history of disease and
the effectiveness of interventions in modifying health outcomes are critical. In addition, if the clinical validity of an early recognition algorithm is not sufficiently sensitive, cases will be missed; if the algorithm is not specific enough, too
many persons will be referred for testing. Proposed algorithms therefore need to be assessed for analytic validity (e.g., comparing the number and type of infections reported by patients to the documentation in the medical record), clinical validity
(e.g., determining the proportion of persons with specific symptoms who have or do not have a PI disease), and clinical utility (e.g., determining whether early detection of a specific disorder affects long-term outcomes and is cost-effective).
The limited experience with symptom-based screening methods for a group of diverse disorders demonstrates the
challenges in establishing clinical algorithms that can be
applied readily in busy clinical practices with accuracy and efficiency
(43,95). Findings indicate that clinical algorithms vary in their analytic and clinical validity, especially depending on the age of the population. Therefore, algorithms must be refined to improve sensitivity and avoid missed cases and to increase specificity
to
reduce costs associated with the immunologic workup of unaffected children and adults. New practice parameters,
including information related to diagnosis and treatment, are in development, and physicians need to be made aware of these to assist in the early identification and management of these patients (L. Kobrynski, M.D., Emory University, Atlanta, Georgia,
personal communication, 2003).
Workshop Recommendations for Action. Different approaches
for early clinical recognition have been used in
clinical settings, but none have been systematically evaluated. Workshop recommendations for early clinical recognition are as follows:
Collect data related to the effect of early interventions on morbidity and mortality associated with PI diseases.
Identify a group of diseases that can benefit from using an early clinical recognition algorithm. Possibilities
include SCID, XLA, CVID, CGD, and WAS.
Establish a working group to create a system of clinical algorithms for early clinical recognition of PI diseases.
The working group should include primary-care physicians. Possible early-recognition tools are scoring systems, lists
of warning signs, questionnaires, or alert bulletins.
Select target audiences and adjust the early-recognition tools for each audience.
Before widespread application of the algorithms, evaluate the usefulness and accuracy of early clinical signs and
symptoms and initial laboratory tests for early recognition of PI diseases. Explore existing databases to test
proposed algorithms.
Report on the effectiveness of the tools among the original target audiences and amend the tools as indicated.
Evaluate the usefulness of family history in recognizing single-gene disorders early.
Conduct collaborative studies among clinical centers to examine the natural history of selected PI diseases.
Conduct research regarding impediments to access to treatment and case management.
Conduct needs assessments related to timely diagnosis, access to treatment, and ongoing care.
Newborn Screening
Certain severe PI diseases become apparent early in life, with only a short asymptomatic period after birth. Without
an effective intervention, the majority result in irreversible complications and death before the end of the first year of life. The most useful method for improving the outcomes of diseases with such a narrow window for detection and intervention
might be population-based newborn screening (NBS).
Existing Newborn Screening Programs. NBS programs began in the 1960s with the development of an accurate
and sensitive test for PKU, an inherited disorder of metabolism
(96). Children affected with PKU are unable to metabolize
the amino acid phenylalanine. If untreated, affected children will be severely mentally retarded and experience other neurologic symptoms. However, dietary therapy started soon
after birth will reduce symptoms and allow affected children to develop normally. The average incidence of PKU is approximately 1/16,000 births.
The PKU assay uses a dried blood spot (DBS) specimen. Blood is collected from the heel of an infant 1--2 days after birth. The heel is pricked, and a few drops of blood are spotted onto a filter paper card, dried, and sent to a state or
regional public health laboratory. Small filter-paper disks containing dried blood are punched from the specimens and used to test the newborn for PKU and other disorders. This simple, easily transported, and inexpensive specimen-collection method has led
to development of population-based screening of newborns throughout the world
(41,97--99). Babies in the United States
are screened for 4--30 different metabolic, hematologic, and endocrinologic disorders within a few days of birth. All of these tests are performed by using DBS specimens. As a population-based public health activity, NBS programs are the responsibility of state public health agencies and operate under policies determined at the state level, although laboratory screening might be contracted to other states or to academic or private laboratories
(97,100).
Newborn Screening Quality Assurance Program. CDC's Newborn Screening Quality Assurance Program
(NSQAP) produces, certifies, and distributes DBS materials for external quality control and performance surveillance to help NBS laboratories evaluate and improve the quality of their testing and to foster standardization of NBS services (101). Approximately 250 national and international screening laboratories from
45 countries participate in the quality assurance program. NSQAP recently added quality assurance materials for disorders detected by tandem mass spectrometry
(102,103) and CF (101).
Principles for Evaluating Evidence for Newborn
Screening. Guidelines for NBS programs are linked to ethical, legal,
and social considerations and are based on the premise that screening should be conducted only when science and technology can serve both the individual person and the public good. Certain landmark reports
(47,98,104) identify criteria for
population-
based NBS programs. The criteria typically follow standard principles of population screening developed in 1968 (105). These principles emphasize the
importance of a specific condition to public health;
availability of an effective screening test;
availability of diagnosis and treatment;
existence of a recognizable latent or early symptomatic phase for the condition and an adequately understood
natural history;
an agreed upon policy regarding whom to treat;
a balance between screening costs and health expenditures; and
availability of case-finding capabilities.
These criteria have been discussed and modified multiple times
(64,100). With the advent of new testing technologies,
the criteria and corresponding evidence and ethical problems are being revisited at the state and national levels
(64).
Newborn Screening and SCID. Among PI diseases, SCID is a candidate for development of an NBS protocol. SCID
is characterized by profound deficiencies of T- and
B-cell function and is usually lethal during infancy without
successful immune reconstitution, ideally during the first months of life
(15,16,19).
Efficacy of Early Identification and
Treatment. Research indicates that infants with SCID who receive
hematopoietic stem-cell transplants from related donors in the first 3.5 months of life have approximately 95% chance of survival,
compared with a survival rate of 76% for infants receiving this treatment after 3.5 months
(27). Infants who received stem cell transplants during the first 28 days of life demonstrated higher levels of T-cell reconstitution and thymic output than did those who received a transplant later; updated survival estimates were 95% (N = 21/22) for infants receiving transplants during
the first 28 days, compared with 74% (N = 71/96) for infants receiving transplants after the neonatal period
(19). An analysis of registry data for 475 SCID patients from 37 centers in 18 European countries reported that long-term survival among
patients who received stem-cell transplants has improved, probably because of more
effective prevention of complications
(106). Differences were identified by SCID phenotype, with poorer outcomes
occurring among SCID patients without B cells than among those with B cells. Immune reconstitution using gene therapy in clinical trials has also been achieved for forms of SCID (14,17,18,20,52); however, as discussed previously, the unexpected complication of T-cell leukemia occurred among
2 of 10 children receiving therapy for
IL2RG SCID (53--55). Similar types of therapy are promising for
other immunodeficiencies (26).
The need to identify at birth children with SCID, as evidenced from clinical studies, permits time to institute therapies
for immune reconstitution before the onset of opportunistic and other infections associated with negative outcomes. SCID meets certain traditional criteria for NBS, as follows
(105):
SCID is fatal during infancy without immune reconstitution.
A short asymptomatic period exists after birth.
Effective treatments are available.
Early intervention improves outcome.
Profound deficiencies of cellular and humoral immunity might be detectable with screening tests.
Development and Evaluation of Screening
Tests. Data regarding the analytic and clinical validity of the screening tests
are critical in considering an NBS program. One study, which was conducted in New York state in the 1970s, assessed
the effectiveness of a DBS screening test for ADA deficiency based on ADA enzyme activity
(107,108). This led to the detection of 12 partially ADA-deficient patients (i.e., persons whose erythrocytes lacked ADA but who had substantial ADA in other cell types and who were clinically and immunologically normal)
(109), but no cases of ADA SCID were
detected. However, because of variability in the tests used, two patients with ADA SCID were missed at one hospital. Data regarding
genotype-phenotype correlation are now accumulating for ADA deficiency and is important to consider in NBS
(110). The majority of ADA-deficient patients have SCID, but in 15%--20% of these, the condition is diagnosed late in childhood or in
adulthood with more variable immunodeficiency; normal persons with partial ADA deficiency also have been identified
(111).
Identification of SCID at birth will require developing a high-throughput screening test. Data indicate that a T-cell
count might be an effective screening tool. The phenotypic hallmark of SCID is profound T-cell lymphopenia, with
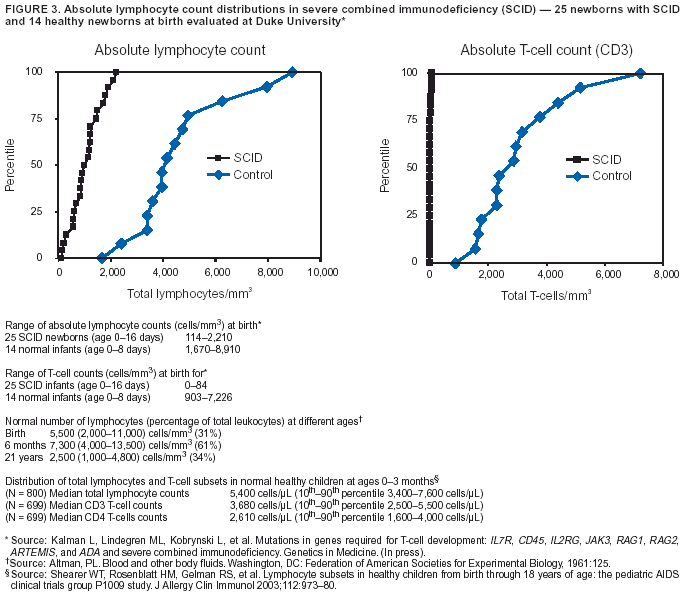
counts substantially below the first percentile of normal; transplacental maternal T-cell engraftment might cause this number to be higher only in a limited number of cases. Compared with healthy infants, whose total lymphocyte counts at birth are
2,000--
11,000 cells/µL (112), counts in SCID patients are usually <1,500--2,000 cells/µL (Figure 3). CD3+ T-cell counts in infants with SCID are typically <500 cells/µL (normal: 3,000--6,500 cells/µL) (15,16,28,113). In a study of a large urban,
primarily minority cohort of 800 healthy children, median total lymphocyte counts at ages 0--3 months were 5,400 cells/µL (10th --90th percentile, 3,400--7,600 cells/µL); median
CD3+ T-cell counts were 3,680 cells/µL
(10th--90th percentile, 2,500--5,500 cells/µL); and CD4+ T-cells were 2,610 cells/µL (10th--90th percentile, 1,600--4,000 cells/µL) (114).
Development of a DBS-based high-throughput test for
T-cell lymphopenia will make possible integration of screening
for SCID into the existing NBS system. Screening tests might detect markers on mummified T-cells (and other
leukocytes) present on DBS. Multiple types of soluble T-cell--specific biomarkers that theoretically can be recovered from DBS have been indicated as potential surrogates for a T-cell count. One such biomarker is the family of cell-membrane antigens unique to T-cells, most notably CD3, CD4, and CD8. Measurements of these T-cell markers from DBS might be possible by
using antibody-based detection assays (115). Another potential biomarker is the circular DNA removed when T-cell--receptor variable genes rearrange during T-cell development. These molecules are called T-cell antigen receptor excision circles (TRECs) (19,116). Detection and quantitation of TRECs from DBS should be possible by using PCR amplification (117). TRECs, located in recently formed T cells, should be abundant in normal newborns but absent in newborns with
SCID. Quantitation of TRECs from NBS with high-throughput
application has not been developed.
Total lymphocyte counts, as obtained in a complete blood count, also have been proposed as a screen for lymphopenia. However, because affected newborns often have increased
B-cell counts that cause an approximate 20% overlap with
the normal lymphocyte distributions, this approach can potentially miss cases of SCID and require supplemental testing
for certain normal newborns (19). Detection of all cases will require enumeration of total lymphocyte counts with a manual differential and subsequent subset analysis by using flow cytometry, neither of which can be performed on DBS specimens. Detection of specific DNA sequences from DBS is also possible. However, although genomic DNA-based tests to detect the disease-causing alleles can be developed on the basis of the detection of one or a limited number of specific mutations, the number and wide spectrum of molecular defects and lack of data regarding genotype-phenotype
relations that can cause SCID currently precludes development of a
specific DNA test.
Evaluation of Newborn Screening. In addition to developing a screening test, other steps need to be taken before routine screening of newborns for SCID can be considered. These include
determining the analytical validity of the proposed assay;
developing a standardized case definition of the disorder;
developing effective follow-up protocols for
screen-positive infants;
identifying treatment centers;
conducting pilot testing to assess the assay's clinical validity, clinical utility, outcomes, and costs;
determining cost-benefit; and
assessing ethical, legal, and social implications.
The possibility of detecting lymphopenia caused by other
genetic causes or HIV infection also needs to be
considered. Although children with these conditions do not have SCID, any child identified with severe lymphopenia requires
further evaluation. By testing all infants, children with a fatal but treatable disease can be identified and treated, and valuable information can be obtained regarding the incidence of these disorders in the population and the frequency of different mutations among affected persons and in the population.
In considering SCID as a possible addition to state newborn screening, evidence-based criteria should be used but
might require re-examination in terms of weighting of different criteria. For example, the question of whether a condition is a key public health problem often is decided on the basis of prevalence. Such disorders as SCID with a prevalence of perhaps 1/100,000 might not be considered a critical public health concern by everyone. Cost concerns (i.e., cost-effectiveness or
cost-benefit of proposed screening tests) are also important and need to be considered systematically. Detection of a disorder with a low prevalence might be more cost-effective than detection of a much more common disorder, depending on the severity
of the health outcomes, effectiveness of interventions, and cost of screening and treatment
(118). Economic analysis is a way of systematically integrating and evaluating multiple screening criteria. State newborn screening advisory committees should consider this more objective process
(119).
Workshop Recommendations for Action. Workshop recommendations
for NBS are as follows:
Determine the feasibility of NBS for SCID.
Establish partnerships among investigators and CDC laboratory personnel to develop assays to measure
T-cell lymphocytes from DBS.
Establish partnerships among investigators and CDC laboratory personnel to validate methods to measure
T-cell lymphocytes or TRECs from DBS. Validation methods can include blinded comparisons of T-cell counts by using the proposed assays from DBS, with a manual differential
count from cord blood samples as the benchmark.
Collaborate with partners to review data regarding
population-based normal ranges of T-cells,
CD4+ cells, and TRECS at birth.
Pilot test a validated assay. Integrate the proposed assays into an existing NBS panel on an investigational basis with Institutional Review Board (IRB) approval. Demonstrate adequate follow-up capacity and ability to
ensure access to treatment without financial barriers.
After pilot testing has demonstrated that NBS for T-cell lymphopenia can
be performed with an extremely high degree of accuracy at acceptable cost and that follow-up services and treatment can
be provided to all affected children identified through screening, a national-level body might recommend that states
include this test in the standard NBS panel. Each state should have an advisory committee to consider such a recommendation.
Evaluation of Screening and Diagnostic Tests
Genetic Tests and PI Diseases. Advances in molecular
biology and genetic technology have facilitated localization
of disease genes and identification of disease-causing mutations, allowing for more rapid development of new genetic tests.
PI diseases are among the approximately 800 health conditions for which genetic tests are available in clinical practice
(120,121). As the genetic defects associated with PI diseases continue to be discovered, more genetic tests will become available for clinical diagnosis, carrier detection, prenatal diagnosis, and disease management
(13,45,122).
The genetic aspects of PI diseases and their implications for diagnosis and patient management have been
extensively reviewed (22). Mutation detection is the most reliable diagnostic method
(45). However, because of the substantial number
of mutations across the spectrum of genes that characterize immunodeficiency, targeting one or a limited number of mutations
is inappropriate. Methodologically, DNA-based detection involves different molecular techniques, although DNA sequencing
is the usual diagnostic method. Evaluation of mRNA or protein also can be used because absent or low levels of specific
mRNA or protein are diagnostic for certain PI diseases. Finally, in conjunction with a family history, clinical and laboratory findings in certain X-linked disorders can also provide a diagnosis.
As tools for the diagnosis and screening of PI diseases evolve, defining and pursuing measures that will ensure their safe and effective use become increasingly critical. Genetic testing in the United States has developed successfully, providing
options for avoiding, preventing, and treating inherited disorders. Nonetheless, application of genetic tests is increasing in clinical and public health practice. Concerns related to rapid commercialization of genetic tests are complex and controversial.
Appropriate use of tests, quality of laboratory testing, direct-to-consumer marketing, and the potential for discrimination
and stigmatization call for public health leadership. Such leadership is needed to protect the public from inappropriate testing
and to ensure that tests are properly evaluated and integrated into medical and public health practice
(47,56).
Evaluation of Genetic Tests. In 1999, the National Institutes of Health (NIH)-U.S. Department of
Energy Task Force on Genetic Testing published recommendations to promote safe and effective genetic testing
(47). The Task Force recognized the need to evaluate genetic tests in population-based settings before their use in clinical practice. To ensure the appropriate level
of review, the panel recommended that genetic tests be evaluated according to three criteria, analytic validity, clinical validity,
and clinical utility. Systematic assessment based on these measures provides data to determine whether a genetic test
being considered for use in population-based screening or clinical diagnosis is safe and effective as the technology moves from research to
clinical settings (123,124). The criteria also can be applied to screening tests and clinical algorithms.
Analytic validity is the ability of a test to measure the analyte of interest. In the case of a genetic test, analytic validity
refers to the ability of the test to classify the genotype or analyte related to the genotype
(125). The four main elements of analytic validity are analytic sensitivity, analytic specificity, laboratory quality control, and assay robustness. However, an analytically valid test is useful only if it helps to diagnose or predict disease (i.e., the test must also be clinically valid)
(125). Clinical validity is the accuracy with which a test predicts a particular clinical outcome. It reflects both the sensitivity of the test ---
the proportion of affected persons with a positive test --- and specificity of the test, penetrance of the mutations identified by
the test, and the prevalence of disease
(123,124). Penetrance is the proportion of persons with the mutation who develop
the disease. Clinical utility is the usefulness of the test and the value of the information to the person being tested. Clinical
utility
is assessed according to the benefits and risks associated with the test and the ensuing result or interventions. Clinical
utility focuses on health outcomes associated with testing and requires an understanding of the natural history of the disorder.
The Foundation for Blood Research, in collaboration with CDC, has developed a framework for assessing the
availability, quality, and usefulness of data related to genetic tests and testing protocols
(126). This approach, called ACCE (analytic
validity; clinical validity; clinical utility; and ethical, legal, and social implications), derives from the three
evaluation criteria described previously, in addition to a fourth that addresses the safeguards and impediments that should be considered in the context of the others
(126,127). The evaluation process begins only after the clinical disorder and the test setting
(e.g., diagnosis or population screening) have been established. Specific questions (Table 2) help to define the disorder, the setting, and the type of testing and to address ACCE. The first disorder to undergo an ACCE review was CF
(61). Others in progress include hereditary hemochromatosis and breast cancer.
Development and Availability of Genetic Tests. The Task Force has addressed the need to encourage development
and maintenance of tests for rare genetic diseases, establish a comprehensive system to collect data related to rare diseases, and assess the validity of genetic tests for these conditions
(47). Evaluation of genetic tests involves collection and analysis of
data regarding analytic validity, clinical validity, clinical
utility, and other aspects from laboratories and users. However, for
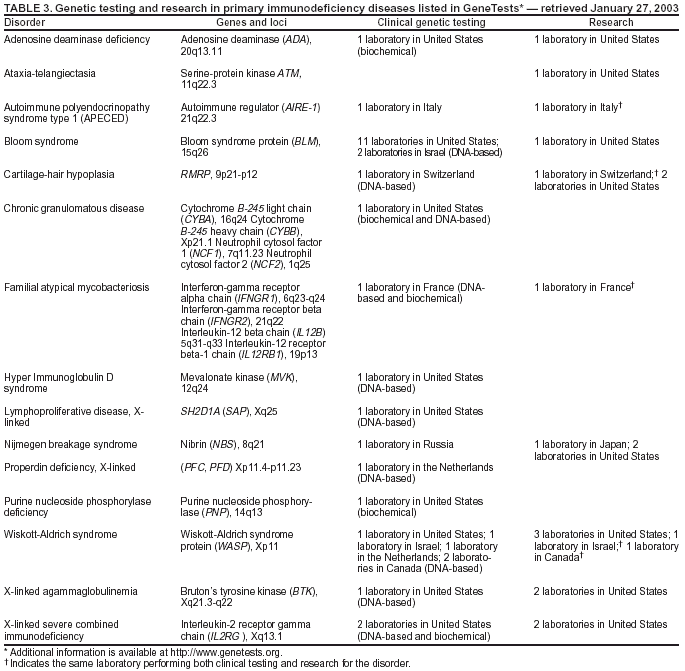
selected PI diseases, genetic testing is available from only a limited number of laboratories, or even only one laboratory, worldwide. Immunodeficiency diseases for which clinical
genetic tests or research testing are available, based on information from
the GeneTests Laboratory Directory (121), are provided in this report (Table 3). The directory lists 11 PI diseases for which clinical genetic tests are offered in only one laboratory; three diseases for which testing is available only outside the United States; and one disease for which testing is available only on a research basis. The limited availability of testing poses challenges for test development and evaluation and presents needs and opportunities for public health
research. Data collection will require a long-term, collaborative effort and a comprehensive, sustainable system to assess the validity and reliability of genetic tests for PI diseases and other rare diseases.
Guidance and criteria for transferring genetic tests from the research and development phase to clinical and public
health practice also are needed. Certain genetic tests were developed in research laboratories and then made available for patient testing. For such rare diseases as PI, a laboratory that primarily conducts research might be the only clinical testing site available. A mechanism needs to be established to enable these laboratories to participate in and contribute to the continuous test evaluation and validation process. Concurrently, criteria need to be developed to guide the transition of genetic testing from research into clinical and public health use.
For certain PI diseases, genetic tests are available only from non-U.S. laboratories (Table 3). The Clinical
Laboratory Improvement Amendments (CLIA) require that U.S. laboratories refer a specimen for testing only to a
CLIA-certified laboratory or a laboratory meeting equivalent requirements as determined by the Center for Medicare and Medicaid Services.* To ensure access to quality genetic testing, a process is needed to evaluate the tests and practices of non-U.S. laboratories that receive test referrals from the United States, determine performance equivalence to CLIA standards, and ensure access to
and availability of testing for rare disorders.
Additional needs include 1) collection of population-based data regarding analytic validity, clinical validity, and
clinical utility for immunologic tests used to diagnose PI diseases
(Figure 2); 2) development of algorithms for use of laboratory
tests and clinical information to increase the likelihood of early clinical diagnosis of PI diseases; and 3) population-based research to evaluate the utility of genetic tests as early diagnostic tools for PI diseases, both as part of NBS programs and for
confirmatory or follow-up diagnosis.
Workshop Recommendations for Action. Recommendations from the workshop include the following:
Evaluate potential genetic tests for their validity, utility, and feasibility as both screening tests and confirmatory or follow-up diagnostics in combination with other tests.
Ensure that CLIA-compliant laboratory testing is accessible, available, and valid for diagnosing rare genetic diseases, including suspected PI diseases, in collaboration with agencies providing oversight for CLIA, NIH Office of
Rare Diseases, CDC, and others.
Support the formation of treatment networks and referral centers to ensure access to diagnosis and care for persons with PI diseases.
Collect data regarding the analytic and clinical validity of molecular tests used for diagnosis and any proposed screening tests.
Review gene databases in the United States and Europe to highlight the availability and possible sources of data regarding the validity and quality of tests.
Identify centers for pilot testing of any proposed screening assays to determine clinical validity, in collaboration with states, CDC, other federal agencies, and other
partners. Integrate any proposed validated assay into an existing NBS
panel on an investigational basis with IRB approval. Demonstrate adequate follow-up capacity and ability to ensure access
to treatment without financial barriers.
Education and Communication
To encourage early recognition of PI diseases, followed by appropriate referral and treatment, primary-care
providers, parents, and other caregivers must be educated regarding the symptoms of PI diseases, resources for referral, and treatment options. The effectiveness of a health communication and education campaign depends on the consistency of the
messages and the coordination of communication strategies to reach targeted audiences among the groups involved in PI
research and education.
Existing Efforts. Multiple agencies and organizations sponsor outreach and educational efforts designed to
increase awareness of PI diseases. NIH, Mt. Sinai Hospital, the Jeffrey Modell Foundation
(128), and IDF (129) have all
targeted proactive outreach efforts to a range of audiences (e.g., health-care providers, patients, families, and teachers), although health-care providers have been the primary focus. Outreach activities and resources include conferences and workshops, Internet-based training and resources, community-based training, distribution of awareness posters, media briefings and news releases, consulting networks, and a visiting professor program. The National Organization for Rare Disorders (NORD) also
provides print and online resources for health-care providers on multiple rare diseases, including PI diseases
(130).
Although these educational efforts have been ongoing for years, outcomes have not been formally evaluated. In
addition, various educational activities or messages have not been coordinated, and consensus has not been developed among the organizations or scientists involved in educational research related to PI diseases. Because the diseases vary in severity, symptoms, etiology, and outcomes, coherent messages regarding groups of PI diseases are difficult to create, and no agreement exists concerning which disorders should be the focus of a health communication campaign. Although educational
efforts should highlight PI diseases that can benefit from and be targeted for early recognition and that have established criteria for early clinical recognition, priorities for educational efforts have yet to be established.
Components of Effective Programs. Effective health communication and education programs should be preceded
by consensus in the scientific community regarding which PI diseases to include in an educational program, the
associated symptoms, and the recommended screening and management steps. To encourage early recognition, education regarding
PI diseases also will need to reach multiple audiences, including the general public, parents, physicians, school nurses, child care providers, and policy makers. Reaching each audience with consistent but targeted messages will require
careful coordination among different agencies.
Attempts to reach primary-care providers, recognized as the front line in the fight against PI diseases, must
overcome multiple barriers. Other diseases with higher prevalence command the attention of physicians. Primary-care providers with heavy caseloads and limited time for continuing education activities probably focus their continuing education efforts
on problems encountered most frequently among primary-care providers. Health-care providers are most likely to attend to
the most prevalent health problems among their patients. The prevalence of PI diseases (individually or collectively) has not been established, although estimates classify them as rare to extremely rare. With such high-prevalence diseases as asthma claiming high priority for providers' attention and concern, focusing on less prevalent health problems might be difficult.
Development of a broad health communication campaign for providers and the public is premature. Research to
determine the prevalence and etiology of PI diseases and the efficacy of early treatment must be completed before effective messages and educational materials for the public and providers can be developed. However, pending delineation of defined
symptoms, disease groups, and treatment recommendations, health communication
efforts still can be useful. Although research has
not yet yielded a defined set of educational goals related to PI diseases, health communication efforts can be used in the interim to increase awareness among scientists and clinicians. Certain health-care providers might be unaware of PI diseases and
research, and researchers might be unaware of opportunities for funding and participation in PI investigations.
Workshop Recommendations for Action. Workshop recommendations include the following:
Target health-care providers and scientists for early-stage communication activities. Increase their awareness of
studies under way, questions motivating research programs, opportunities for participation and funding, and resources.
Use research concerning the outcomes of previous and ongoing educational programs to determine how best to
reach target audiences with information related to PI diseases. Systematically analyze the range of outreach efforts to determine 1) information reach, 2) frequency of message contact, and 3) interaction of messages from different organizations. Use evidence-based outcome assessments to determine awareness, knowledge, and uses of information
from previous education and communication programs.
Convene a working group of health communication specialists to establish campaign goals, audiences, and strategies,
even as research continues and consensus is reached regarding disorders to include in a health communication campaign
and case definitions and clinical recommendations are developed. The working group should
--- determine additional formative research needed to
assess target audiences' awareness, knowledge, and
behaviors related to PI;
--- develop or revise materials that are consistent with
campaign goals;
--- develop additional materials as needed to achieve
campaign goals;
--- pretest materials with target audiences;
--- disseminate messages that are consistent with recommendations from pretesting, and
--- include process and evidence-based outcome evaluations as part of campaign planning.
Conclusion
This report presents a framework for stakeholders and policy makers who will collaborate to define the future of
an emerging and promising field of study that can markedly improve health in persons with PI diseases. The
recommended interventions encompass multiple goals --- helping children, educating clinicians, developing and maintaining awareness of PI diseases, and providing information for policy development and change. Additional efforts are needed to define priorities
in future public health actions and associated costs and benefits. The proposed public health framework is critical for PI
diseases and serves as a model for other genetic disorders that can benefit from early diagnosis and opportunities for interventions
to improve health outcomes.
References
Khoury MJ, Burke W, Thomson, EJ. Genetics and public health: a framework for the integration of human genetics into public health practice.
In: Khoury MJ, Burke W, Thomson, EJ, eds. Genetics and public health in the
21st century: using genetic information to
improve health and prevent disease. New York, NY: Oxford University Press, 2000.
Koplan JP, Fleming DW. Current and future public health challenges. JAMA 2000;284:1696--8.
Little J, Bradley L, Bray MS, et al. Reporting, appraising, and integrating data on genotype prevalence and gene-disease associations. Am
J Epidemiol 2002;156:300--10.
Little J, Khoury MJ, Bradley L, et al. The human genome project is complete. How do we develop a handle for the pump? Am J
Epidemiol 2003;157:667--73.
Khoury MJ. Commentary: epidemiology and the continuum from genetic research to genetic testing. Am J Epidemiol 2002;156:297--9.
Collins FS, Green ED, Guttmacher AE, Guyer MS. A vision for the future of genomics research. Nature 2003;422:835--47.
Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature 2001;409:860--921.
McPherson JD, Marra M, Hillier L, et al. A physical map of the
human genome. Nature 2001;409:934--41.
Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science 2001;291:1304--51.
Khoury MJ. Genetics and genomics in practice: the continuum from genetic disease to genetic information in health and disease. Genet
Med 2003;5:261--8.
Anonymous. Primary immunodeficiency diseases. Report of IUIS scientific committee. International Union of Immunological Societies. Clin
Exp Immunol 1999;118(Suppl 1):1--28.
Chapel H, Geha R, Rosen F, for the IUIS PID Classification Committee. Primary immunodeficiency diseases: an update. Clin Exp
Immunol 2003;132:9--15.
Puck JM. Primary immunodeficiency diseases. JAMA 1997;278:1835--41.
Aiuti A, Slavin S, Aker M, et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning.
Science 2002;296:2410--3.
Buckley RH. Advances in the understanding and treatment of human severe combined immunodeficiency. Immunol Res 2000;22:237--51.
Buckley RH. Primary immunodeficiency diseases due to defects in
lymphocytes. N Engl J Med 2000;343:1313--24.
Cavazzana-Calvo M, Hacein-Bey S, Yates F, de Villartay JP, Le Deist F, Fischer A. Gene therapy of severe combined immunodeficiencies.
J Gene Med 2001;3:201--6.
Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease.
Science 2000;288:669--72.
Myers LA, Patel DD, Puck JM, Buckley RH. Hematopoietic stem cell transplantation for severe combined immunodeficiency in the neonatal
period leads to superior thymic output and improved survival. Blood 2002;99:872--8.
Hacein-Bey-Abina S, Le Deist F, Carlier F, et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy.
N Engl J Med 2002;346:1185--93.
Guttmacher AE, Collins FS. Genomic medicine---a primer. N Engl J Med 2002;347:1512--20.
Smith CIE, Ochs HD, Puck JM. Genetically determined immunodeficiency diseases: a perspective. In: Ochs HD, Smith CIE, Puck JM,
eds. Primary immunodeficiency diseases: a molecular and genetic
approach. New York, NY: Oxford University Press, 1999.
Noroski LM, Shearer, WT. Screening for primary immunodeficiencies in the clinical immunology laboratory. Clin Immunol
Immunopathol 1998;86:237--45.
Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin
Immunol 1999;92:34--48.
Buckley RH, Fischer A. Bone marrow transplantation for primary immunodeficiency diseases. In: Ochs HD, Smith CIE, Puck JM, eds.
Primary immunodeficiency diseases: a molecular and genetic approach. New York, NY: Oxford University Press, 1999;459.
Buckley RH, Schiff SE, Schiff RI, et al. Hematopoietic stem-cell transplantation for the treatment of severe combined immunodeficiency. N Engl
J Med 1999;340:508--16.
Kalman, L, Lindegren, ML, Kobrynski L, et al. Mutations in genes required for T-cell development:
IL7R, CD45, IL2RG,
JAK3, RAG1, RAG2,
ARTEMIS, and ADA and severe combined immunodeficiency. Genetics in Medicine. (In press).
Chinen J, Rosenblatt HM, Smith EO, Shearer WT, Noroski LM. Long-term assessment of T-cell populations in DiGeorge syndrome.
J Allergy Clin Immunol 2003;111:573--9.
Jawad AF, McDonald-Mcginn DM, Zackai E, Sullivan KE. Immunologic features of chromosome 22q11.2 deletion syndrome (DiGeorge
syndrome/velocardiofacial syndrome). J Pediatr 2001;139:715--23.
Baumgart KW, Britton WJ, Kemp A, French M, Roberton D. The spectrum of primary immunodeficiency disorders in Australia.
J Allergy Clin Immunol 1997;100:415--23.
Matamoros Flori N, Mila Llambi J, Espanol Boren T, Raga Borja S, Fontan Casariego G. Primary immunodeficiency syndrome in Spain: first
report of the National Registry in Children and Adults. J Clin Immunol 1997;17:333--9.
Stray-Pedersen A, Abrahamsen TG, Froland SS. Primary immunodeficiency diseases in Norway. J Clin Immunol 2000;20:477--85.
Ryser O, Morrell A, Hitzig WH. Primary immunodeficiencies in Switzerland: first report of the national registry in adults and children.
J Clin Immunol 1988:479--85.
Fasth A. Primary immunodeficiency disorders in Sweden: cases among children, 1974--1979. J Clin Immunol 1982;2:86--92.
Winkelstein JA, Marino MC, Johnston RB Jr, et al. Chronic granulomatous disease. Report on a national registry of 368 patients.
Medicine (Baltimore) 2000;79:155--69.
Clark JA, Callicoat PA, Brenner NA, Bradley CA, Smith DM Jr. Selective
IgA deficiency in blood donors. Am J Clin Pathol 1983;80:210--3.
Immune Deficiency Foundation. The clinical presentation of the primary immunodeficiency diseases (physician's primer). Towson, MD:
Immune Deficiency Foundation, 1992. Available at
http://www.primaryimmune.org/pubs/book_phys/book_phys.htm.
Shearer WT, Paul ME, Smiht CW, Huston DP. Laboratory assessment of immune deficiency disorders. Immunol Allergy Clin N Am
1994;14:265--99.
CDC. Newborn screening for a cystic fibrosis: a paradigm for public health genetics policy development. Proceedings of a 1997 workshop.
MMWR 1997;46(No. RR-16):1--24.
National Institutes of Health Consensus Development Panel. National Institutes of Health Consensus Development Conference
statement: phenylketonuria: screening and management, October 16--18, 2000. Pediatrics 2001;108:972--82.
Puck JM. Genetic aspects of primary immunodeficiencies. In: Ochs HD, Smith CIE, and Puck JM, eds. Primary immunodeficiency
diseases: a molecular and genetic approach. New York, NY: Oxford University Press, 1999.
Kobrynski L. Evaluation of a clinical scoring system for the identification of patients with a possible primary immunodeficiency [Abstract
379]. Presented at the Federation of Clinical Immunology Society Meeting, 2002.
Zhu Q, Watanabe C, Liu T, et al. Wiskott-Aldrich syndrome/X-linked thrombocytopenia: WASP gene mutations, protein expression,
and phenotype. Blood 1997;90:2680--9.
Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group
for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol 1999;93:190--7.
Chapel, HM, Webster, ADB. Assessment of the immune system. In: Ochs HD, Smith CIE, Puck JM, eds. Primary immunodeficiency
diseases: a molecular and genetic approach. New York, NY: Oxford University Press, 1999.
Holtzman NA, Watson MS. Promoting safe and effective genetic testing in the United States: Final report of the task force on genetic testing.
J Child Fam Nurs 1999;2:388--90.
Stiehm, ER. Conventional therapy of immunodeficiency diseases. Ochs HD, Smith CIE, Puck JM. Primary immunodeficiency diseases: a
molecular and genetic approach. New York, NY: Oxford University Press, 1999.
Chapel HM for the Consensus Panel for the Diagnosis and Management of Primary Antibody Deficiencies. Consensus on diagnosis
and management of primary antibody deficiencies. BMJ 1994;308:581--5.
Hershfield MS. PEG-ADA: an alternative to haploidentical bone marrow transplantation and an adjunct to gene therapy for adenosine
deaminase deficiency. Hum Mutat 1995;5:107--12.
Haddad E, Landais P, Friedrich W, et al. Long-term immune reconstitution and outcome after HLA-nonidentical T-cell--depleted bone
marrow transplantation for severe combined immunodeficiency: a
European retrospective study of 116 patients. Blood 1998;91:3646--53.
Fischer A, Landais P, Friedrich W, et al. European experience of bone-marrow transplantation for severe combined
immunodeficiency. Lancet 1990;336:850--4.
Buckley RH. Gene therapy for SCID---a complication after remarkable progress. Lancet 2002;360:1185--6.
Buckley RH. Transplantation immunology: organ and bone marrow.
J Allergy Clin Immunol 2003;111(Suppl 2):S733--44.
Hacein-Bey-Abina S, von Kalle C, Schmidt M, et al. A serious adverse event after successful gene therapy for X-linked severe
combined immunodeficiency. N Engl J Med 2003;348:255--6.
CDC. Translating advances in human genetics into public health
action: a strategic plan. Atlanta, GA: US Department of Health and
Human Services, 1997. Available at
http://www.cdc.gov/genomics/about/strategic.htm.
Brown AS, Gwinn M, Cogswell ME, Khoury MJ. Hemochromatosis-associated morbidity in the United States: an analysis of the National
Hospital Discharge Survey, 1979--1997. Genet Med 2001;3:109--11.
McDonnell SM, Witte DL, Cogswell ME, McIntyre R. Strategies to increase detection of hemochromatosis. Ann Intern Med 1998;129:987--92.
Wetterhall SF, Cogswell ME, Kowdley KV. Public health surveillance for hereditary hemochromatosis. Ann Intern Med 1998;129:980--6.
Yang Q, McDonnell SM, Khoury MJ, Cono J, Parrish RG. Hemochromatosis-associated mortality in the United States from 1979 to 1992:
an analysis of multiple-cause mortality data. Ann Intern med 1998;129:946--53.
Foundation for Blood Research. FBR: Foundation for Blood Research [Website]. Scarborough, ME: Foundation for Blood Research, 2003.
Available at http://www.fbr.org.
Cogswell ME, Burke W, McDonnell SM, Franks AL. Screening for hemochromatosis. A public health perspective. Am J Prev Med 1999;16:134--40.
Burke W, Thomson E, Khoury MJ, et al. Hereditary hemochromatosis: gene discovery and its implications for population-based screening.
JAMA 1998;280:172--8.
Khoury MJ, McCabe LL, McCabe ER. Population screening in the age of genomic medicine. N Engl J Med 2003;348:50--8.
CDC. CDC meeting synopsis: applying genetics and public health strategies to primary immunodeficiency diseases. Atlanta, GA: US Department
of Health and Human Services, CDC, 2002. Available at
http://www.cdc.gov/genomics/info/conference/PIsynop.htm.
Thacker SB, Stroup DF. Future directions for comprehensive public health surveillance and health information systems in the United States. Am
J Epidemiol 1994;140:383--97.
Teutsch SM. Considerations in planning a surveillance system. Teutch SM, Churchill RE. Principles and practice of public health surveillance.
2nd ed. New York, NY: Oxford University Press, 2000.
Schulman, Ronca, & Bucuvalas, Inc. Primary immune deficiency diseases in America: the first national survey of patients and specialists.
Towson, MD: Immune Deficiency Foundation, 1999. Available at
http://www.primaryimmune.org/pid/patient_survey_publication.pdf.
Lappalainen I, Ollila J, Smith CI, Vihinen M. Registries of immunodeficiency patients and mutations. Hum Mutat 1997;10:261--7.
European Society for Immunodeficiencies. ESID: European Society for Immunodeficiencies [Website]. Hertogenbosch, The Netherlands:
European Society for Immunodeficiencies, 2003. Available at
http://www.esid.org.
Wang SS, FitzSimmons SC, O'Leary LA, Rock MJ, Gwinn ML, Khoury MJ. Early diagnosis of cystic fibrosis in the newborn period and risk
of Pseudomonas aeruginosa acquisition in the first 10 years of life: a registry-based longitudinal study. Pediatrics 2001;107:274--9.
Vihinen M, Lehvasliho H, and Cotton RGH. Immunodeficiency mutation databases. In: Ochs HD, Smith CIE, Puck, JM, eds.
Primary immunodeficiency diseases: a molecular and genetic approach. New York, NY: Oxford University Press, 1999.
Valiaho J, Riikonen P, Vihinen M. Novel immunodeficiency data servers. Immunol Rev 2000;178:177--85.
Valiaho J, webmaster. BTKbase: mutation registry for X-linked agammaglobulinemia . Tampere, Finland: IMT Bioinformatics, 1995. Available
at http://bioinf.uta.fi/BTKbase.
Porter CJ, Talbot CC, Cuticchia AJ. Central mutation databases---a review. Hum Mutat 2000;15:36--44.
Yoon PW, Olney RS, Khoury MJ, Sappenfield WM, Chavez GF, Taylor D. Contribution of birth defects and genetic diseases to
pediatric hospitalizations: a population-based study. Arch Pediatr Adolesc Med 1997;151:1096--103.
Davis RL, Rubanowice D, Shinefield HR, et al. Immunization levels among premature and low-birth--weight infants and risk factors for delayed
up-to-date immunization status. Centers for Disease Control
and Prevention Vaccine Safety Datalink Group. JAMA 1999;282:547--53.
Belay ED, Holman RC, Clarke MJ, et al. The incidence of Kawasaki syndrome in West Coast health maintenance organizations. Pediatr Infect Dis
J 2000;19:828--32.
Black S, Shinefield H, Ray P, et al. Risk of hospitalization because of aseptic meningitis after measles-mumps-rubella vaccination in one- to
two-year-old children: an analysis of the Vaccine Safety Datalink (VSD) Project. Pediatr Infect Dis J 1997;16:500--3.
Rasmussen SA, Yang Q, Friedman J M. Mortality in neurofibromatosis 1: an analysis using U S death certificates. Am J Hum Genet
2001:68:1110--8.
Yang Q, Khoury MJ, Mannino D. Trends and patterns of mortality associated with birth defects and genetic diseases in the United States,
1979--1992: an analysis of Multiple-Cause Mortality Data. Genet Epidemiol 1997;14:493--505.
Halliburton CS, Mannino DM, Olney RS. Cystic fibrosis deaths in the United States from 1979 through 1991. An analysis using
multiple-cause mortality data. Arch Pediatr Adolesc Med 1996;150:1181--5.
CDC. Mortality data, multiple cause-of-death public-use data files. Hyattsville, MD: US Department of Health and Human Services,
CDC, National Center for Health Statistics, 2003. Available at
http://www.cdc.gov/nchs/products/elec_prods/subject/mortmcd.htm.
World Health Organization. International classification of diseases: manual of the international statistical classification of diseases, injuries,
and causes of death. 9th rev. Geneva, Switzerland: World Health Organization, 1977.
World Health Organization. ICD-10: International statistical classification of diseases and related health problems.
10th rev. Geneva, Switzerland: World Health Organization, 1992.
Botto LD, Mastroiacovo P. Surveillance for birth defects and genetic disorders. In: Khoury MJ, Burke W, Thomson EJ, eds. Genetics and
public health in the 21st century: using genetic information to
improve health and prevent disease. New York, NY: Oxford University Press, 2000.
Kobrynski LJ, Lindgren ML, Rasussen SA, Yang, QH. Using U.S. death certificates to analyze trends in mortality from primary
immunodeficiency diseases [Abstract]. Presented at the Federation of Clinical
Immunology Society Meeting, 2002.
Soucie JM, Nuss R, Evatt B, et al. Mortality among males with hemophilia: relations with source of medical care. The Hemophilia
Surveillance System Project Investigators. Blood 2000;96:437--42.
Soucie J, Rickles FR, Evatt BL. Surveillance for hemophilia and inherited hematologic disorders. In: Khoury MJ, Burk W, Thomson EJ,
eds. Genetics and public health in the
21st century. New York, NY: Oxford University Press, 2000.
CDC. Report on the Universal Data Collection Program (UDC):
includes data collected from May 1998 through September 2001.
Atlanta, GA: US Department of Health and Human Services, CDC, 2002. Available at
http://www.cdc.gov/ncidod/dastlr/Hematology/udc0102.pdf.
Busse PJ, Razvi S, Cunningham-Rundles C. Efficacy of intravenous immunoglobulin in the prevention of pneumonia in patients with
common variable immunodeficiency. J Allergy Clin Immunol 2002;109:1001--4.
Conley ME, Howard V. Clinical findings leading to the diagnosis of X-linked agammaglobulinemia. J Pediatr 2002;141:566--71.
Stadtmauer G, Cunningham-Rundles C. Outcome analysis and cost assessment in immunologic disorders. JAMA 1997;278:2018--23.
Lyall EG, Eden OB, Dixon R, Sutherland R, Thomson A. Assessment of a clinical scoring system for detection of immunodeficiency in
children with recurrent infections. Pediatr Infect Dis J 1991;10:673--6.
Guthrie R, Susi A. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics
1963;32:338--43.
Levy HL, Albers S. Genetic screening of newborns. Annu Rev Genomics Hum Genet 2000;1:139--77.
National Academy of Sciences. National Research Council and Committee for the Study of Inborn Errors of Metabolism. Washington,
DC: National Academy of Sciences, 1975.
Green A, Pollitt RJ. Population newborn screening for inherited metabolic disease: current UK perspectives. J Inherit Metab Dis 1999;22:572--9.
Therrell BL Jr. Minireview: U.S. newborn screening policy dilemmas for the twenty-first century. Mol Genet Metab 2001;74:64--74.
CDC. Newborn screening: preventing mental retardation and other serious health conditions among children [Website]. Atlanta, GA:
US Department of Health and Human Services, CDC, National Center for Environmental Health, 2003. Available at
http://www.cdc.gov/nceh/dls/factsheets/newborn_screening.htm.
CDC. Newborn Screening Quality Assurance Program: 2001 annual summary report. Atlanta, GA: US Department of Health
and Human Services, CDC, 2002. Available at
http://www.cdc.gov/nceh/dls/newborn_screening.htm.
CDC. Newborn Screening Quality Assurance Program: 2001 tandem mass spectrometry annual summary report. Atlanta, GA: US Department
of Health and Human Services, CDC, 2002. Available at
http://www.cdc.gov./nceh/dls/newborn_screening.htm.
Institute of Medicine. Assessing genetic risks: implications for health and social policy. Washington, DC: National Academy Press, 1994.
Wilson JMG, Junger F. Principles and practice of screening for disease. Geneva, Switzerland: World Health Organization, 1968. Public
health papers no. 34.
Antoine C, Muller S, Cant A, et al. Long-term survival and transplantation of haemopoietic stem cells for immunodeficiencies:
report of the European experience 1968--99. Lancet 2003;361:553--60.
Moore EC, Meuwissen HJ. Screening for ADA deficiency. J Pediatr 1974;85:802--4.
Hodes RM. Testing newborns for adenosine deaminase deficiency not cost effective. N Engl J Med 1981;305:1530.
Hirschhorn R. Adenosine deaminase deficiency. Immunodefic Rev 1990;2:175--98.
Arredondo-Vega FX, Santisteban I, Daniels S, Toutain S, Hershfield MS. Adenosine deaminase deficiency: genotype-phenotype correlations
based on expressed activity of 29 mutant alleles. Am J Hum Genet 1998;63:1049--59.
Jenkins T, Rabson AR, Nurse GT, Lane AB. Deficiency of adenosine deaminase not associated with severe combined immunodeficiency.
J Pediatr 1976;89:732--6.
Altman P. Blood and other body fluids. Washington DC: Federation of American Societies for Experimental Biology, 1961;125.
Elder ME. T-cell immunodeficiencies. Pediatr Clin North Am 2000;47:1253--74.
Shearer WT, Rosenblatt HM, Gelman RS, et al. Lymphocyte subsets in healthy children from birth through 18 years of age: the pediatric
AIDS clinical trials group P1009 study. J Allergy Clin Immunol 2003;112:973--80.
Mwaba P, Cassol S, Pilon R, et al. Use of dried blood spots to measure
CD4+ lymphocyte counts in HIV-1--infected patients.
Lancet 2003;362:1459--60.
Hazenberg MD, Verschuren MC, Hamann D, Miedema F, van Dongen JJ. T cell receptor excision circles as markers for recent thymic
emigrants: basic aspects, technical approach, and guidelines for interpretation. J Mol Med 2001;79:631--40.
Chan K, Chinen J, Puck J. Development of population based newborn screening for severe combined immunodeficiency. Clin
Immunol 2003;(Suppl):S104.
Lord J, Thomason MJ, Littlejohns P, et al. Secondary analysis of economic data: a review of cost-benefit studies of neonatal screening
for phenylketonuria. J Epidemiol Community Health 1999;53:179--86.
Grosse S, Gwinn M. Assisting states in assessing newborn screening options. Public Health Rep 2001;116:169--72.
Yoon PW, Chen B, Faucett A, et al. Public health impact of genetic tests at the end of the
20th century. Genet Med 2001;3:405--10.
Children's Health System and the University of Washington. Genetests: medical genetics information resource [Online database].
Seattle, Washington: Children's Health System and the University of Washington, 2003. Available at
http://www.genetests.org.
Fanos JH, Davis J, Puck JM. Sib understanding of genetics and
attitudes toward carrier testing for X-linked severe combined
immunodeficiency. Am J Med Genet 2001;98:46--56.
Burke W. Genetic testing. N Engl J Med 2002;347:1867--75.
Burke W, Atkins D, Gwinn M, et al. Genetic test evaluation: information needs of clinicians, policy makers, and the public. Am J Epidemiol 2002;156:311--8.
Secretary's Advisory Committee on Genetic Testing (SACGT),
National Institutes of Health. A public consultation on oversight of genetic
tests December 1, 1999--January 31, 2000: summary. Bethesda, MD: National Institutes of Health, 2000.
Haddow, JE, Palowmaki, GE. ACCE: a model process for evaluating data on emerging genetic tests. In: Khoury MJ, Little J, Burke, W,
eds. Human genomic epidemiology. New York, NY: Oxford University Press, 2003.
Wald N, Cuckle H. Reporting the assessment of screening and diagnostic tests. Br J Obstet Gynaecol 1989;96:389--96.
Jeffrey Modell Foundation. Jeffrey Modell Foundation [Website]. New York, NY: The Jeffrey Modell Foundation, 2003. Available at
http://www.jmfworld.com.
129. Immune Deficiency Foundation. Immune Deficiency Foundation [Website]. Towson, MD: Immune Deficiency Foundation, 2003. Available
at http://www.primaryimmune.org.
130. National Organization for Rare Disorders (NORD). The National Organization for Rare Disorders (NORD) [Website]. Washington, DC,
2003. Available at http://www.rarediseases.org.
* 42 CFR§493.1242(c).
Terms and Abbreviations Used in This Report*
ACCE analytic validity; clinical validity; clinical utility; and ethical, legal, and social implications
ADA adenosine deaminase gene
AICDA activation-induced cytidine deaminase gene
allele alternative form of a gene that exists at a specific gene location (locus) on a chromosome
analyte substance measured by a laboratory test
APECED autoimmune polyendocrinopathy with candidiasis and ectodermal dysplasia
autosome nuclear chromosomes other than sex chromosomes; the diploid human genome consists
of 46 chromosomes: 22 pairs of autosomes, and one pair of sex chromosomes (the X and
Y chromosomes)
autosomal dominant abnormal gene on one of the autosomal chromosomes from either parent, transmission of
which can cause a particular trait or disorder
autosomal recessive abnormal gene on one of the autosomal chromosomes from each parent, transmission of
both abnormal genes is required to cause a particular trait or disorder
B cell antibody-producing lymphocyte; a type of white blood cell
birth defect defect present at birth, whether caused by mutant genes or by prenatal events that are not genetic
BTK Bruton's tyrosine kinase gene
CF cystic fibrosis
CFF Cystic Fibrosis Foundation
CGD chronic granulomatous disease
chromosome one of the thread-like structures in the cell nucleus; consists of chromatin and carries
genetic information (DNA); human cells normally contain 46 chromosomes (23 pairs)
CLIA Clinical Laboratory Improvement Amendments
codon three-base sequence of DNA or RNA that specifies an amino acid
complement a set of serum proteins that binds antigen-antibody complexes to kill microorganisms
CVID common variable immunodeficiency
DBS dried blood spot
deletion particular kind of mutation; loss of a piece of DNA from a chromosome
DNA deoxyribonucleic acid
EDA-ID ectodermal dysplasia associated with immune deficiency
enzyme protein that facilitates a specific biochemical reaction
ESID European Society for Immunodeficiencies
exon protein-coding DNA sequence of a gene
gene functional and physical unit of heredity, consisting of a segment of DNA arranged linearly
along a chromosome; the majority of genes contain the information for making a specific
protein leading to a particular characteristic or function
gene product biochemical material, either RNA or protein, resulting from expression of a gene
gene therapy treatment of a genetic disorder by replacing, supplementing, or manipulating nonfunctional genes with normal genes
genetic marker landmark for a target gene, either a detectable trait that is inherited with the gene or a distinctive segment of DNA
genetic testing examining a sample of blood or other body fluid or tissue for biochemical, chromosomal,
or genetic markers that indicate the presence or absence of genetic disease
genome complete DNA sequence, containing all genetic information and supporting proteins, in
the chromosomes of a person or species
genomics study of the functions and interactions of all the genes in the genome, including
their interactions with environmental factors
genotype a person's genetic makeup, specifically the alleles present at specific gene loci
genotype/phenotypecorrelation
association between the presence of a certain mutation or mutations (genotype) and the resulting physical trait, abnormality, or pattern of abnormalities (phenotype)
HIV human immunodeficiency virus
HTCs hemophilia treatment centers
Human Genome Project international research project to map each human gene and to completely sequence human DNA
IDF Immune Deficiency Foundation
Ig immunoglobulin
ILR2Ginterleukin 2 receptor gamma gene
incidence number or proportion of new cases of a specified condition among a population during
a specified period
inherited transmitted through genes from parents to offspring
insertion type of mutation in which a DNA sequence is inserted into a gene, disrupting the
normal structure and function of that gene
IRB Institutional Review Board
IVIG intravenous immunoglobulin
JAK3 Janus-associated kinase 3 gene
LAD leukocyte adhesion defect
locus position on a chromosome where a specific gene is located
microarray technology methods for measuring expression of multiple genes simultaneously under specific
conditions relative to baseline (i.e., up regulation or down regulation)
missense a genetic mutation that alters the amino acids in the protein product of a gene
mRNA messenger RNA
mutation permanent heritable change in the molecular sequence of a gene
NADPH nicotinamide-adenine dinucleotide phosphate
NBS newborn screening
Negative predictive value likelihood that a person with a negative test result is actually not affected by the disease
NHGRI National Human Genome Research Institute
NIH National Institutes of Health
nonsense a genetic mutation in single base-pair substitution in DNA resulting in premature stop codons in the genetic code
NORD National Organization for Rare Disorders
NSQAP Newborn Screening Quality Assurance Program
PCR polymerase chain reaction
penetrance frequency with which a genotype manifests itself in a specific phenotype
phenotype clinical presentation or expression of a specific gene or genes, environmental factors, or both
PI primary immunodeficiency
PKU phenylketonuria
positive predictive value likelihood that a person with a positive test result is actually affected by the disease
prevalence number or proportion of existing cases of a specified condition in a population
RAG recombination-activating gene
regulatory (gene) a genetic mutation that affects aspects of gene expression
RNA ribonucleic acid
SCID severe combined immunodeficiency
screening testing on a population basis to identify persons at risk for developing specific disorders
sensitivity frequency with which a test yields a positive result when the abnormality or disease in question
is actually present in the person being tested
sequencing process by which the nucleotide sequence is determined for a segment of DNA
sex chromosome the X and Y chromosomes
single-gene disorder a disorder caused by one or a pair of mutant alleles at a single locus
specificity frequency with which a test yields a negative result when the abnormality or disease in question is not present in the person being tested
splice site a genetic mutation that can lead to frameshift mutations
T cell a white blood cell or lymphocyte that develops in the thymus and mediates cellular
immune responses
TRECs T-cell antigen receptor excision circles
UDC universal data collection
WAS Wiskott-Aldrich syndrome
X-linked recessive genes transmitted on the X chromosome
Applying Genetic and Public Health Strategies to Primary Immunodeficiency
Consultants: Francisco Bonilla, M.D., American Academy of Asthma, Allergy, and Immunology/Children's Hospital, Boston, Massachusetts; Barbara Brenner, Dr.P.H. Mt. Sinai Hospital, New York, New York; Rebecca H. Buckley, M.D., Duke University Medical Center, Durham, North
Carolina; Nancye Buelow, Genetic Alliance, Clyde, North Carolina; Preston Campbell, M.D., Cystic Fibrosis Foundation, Bethesda, Maryland; Elaine Collier, M.D., National Institutes of Health, Bethesda, Maryland; Anne Marie Comeau, Ph.D., New England Newborn Screening Program, Jamaica Plain, Massachusetts; Mary Ellen Conley, M.D., St. Jude Children's Research Hospital, Memphis, Tennessee; Chris Cunniff, M.D., American Academy of Pediatrics/University of Arizona College of Medicine, Tucson, Arizona; Charlotte Cunningham-Rundles, M.D., Ph.D., Mt. Sinai School of Medicine, New York, New York; Lyle Dennis, Cavarocchi Ruscio Dennis (CRD) Associates, Washington, D.C.; Roger Eaton, Ph.D., New England
Newborn Screening Program, Jamaica Plain, Massachusetts; Jonathan Goldsmith, M.D., Immune Deficiency Foundation, Towson, Maryland; Nancy S. Green, M.D., March of Dimes, White Plains, New York. Edward Gruson, National Organization for Rare Disorders, Fairfield, Connecticut; James Haddow, M.D., Foundation for Blood Research, Scarborough, Maine; Celine Hanson, M.D., Texas Department of Health, Austin, Texas; Michael Hershfield, M.D., Duke University Medical Center, Durham, North Carolina; Richard Hong, M.D., University of Vermont, Burlington, Vermont; Lisa
Kobrynski, M.D., Emory University, Atlanta, Georgia; Allan Lock, D.V.M., National Institutes of Health, Bethesda, Maryland; John Meaney, Ph.D., University of Arizona Health Science Center, Tucson, Arizona; Fred Modell and Vicki Modell, The Jeffrey Modell Foundation, New York, New York; Thomas
L. Moran, Immune Deficiency Foundation, Towson, Maryland; Andre J. Nahmias, M.D., Emory University, Atlanta, Georgia; Hans D. Ochs,
M.D., University of Washington School of Medicine, Seattle, Washington; James M. Oleske, M.D., New Jersey Medical School, Newark, New Jersey; Mary
E. Paul, M.D., Texas Children's Hospital, Houston, Texas; Jennifer M. Puck, M.D., National Institutes of Health, Bethesda, Maryland; Michele Lloyd-Puryear, M.D., Ph.D., Health Resources and Services Administration, Rockville, Maryland; Chaim Roifman, M.D., The Hospital for Sick
Children, Toronto, Ontario, Canada; John Salamone, Advisory Committee on Immunization Practice/National Italian American Foundation, Washington, D.C.; William T. Shearer, M.D., Ph.D., Clinical Immunology Society/Baylor College of Medicine, Houston, Texas; Priscilla Short, M.D., American Medical Association, Chicago, Illinois; C.I. Edvard Smith, M.D., Ph.Dm. European Society for Immunodeficiencies, Karolinska Institutet, Huddinge, Sweden;
Richard Stiehm, M.D., University of California at Los Angeles, Los Angeles, California; Brad Therrell, Ph.D., National Newborn Screening
and Genetics Resource Center, Austin, Texas; Tracy Trotter, M.D., American Academy of Pediatrics, San Ramon, California; Mike Watson, Ph.D., American College of Medical Genetics, Bethesda, Maryland; and Jerry Winkelstein, M.D., Immune Deficiency Foundation/Johns Hopkins Hospital, Baltimore, Maryland.
CDC Staff: Richard J. Jackson, M.D., Timothy G. Baker, M.P.H., Scott Grosse, Ph.D., Marta Gwinn, M.D., Muin Khoury, M.D., Ph.D., Mary
Lou Lindegren, M.D., Marifran Mattson, Ph.D., Robert F. Vogt, Jr, Ph.D., and Paula Yoon, Sc.D., National Center for Environmental Health; José Cordero, M.D., Coleen Boyle, Ph.D., Amanda Brown, Ph.D., Larry Edmonds, M.S.P.H., Katherine Lyon-Daniel, Ph.D., Cynthia A. Moore, M.D., Ph.D.,
and Sonja Rasmussen, M.D., National Center on Birth Defects and Developmental Disabilities; Sherry Orloff, M.P.H., National Center for HIV, STD, and TB Prevention; Sally Crudder, M.P.H., Steve McDougal, M.D., Mike Soucie, Ph.D., and Tom Spira, M.D., National Center for Infectious Diseases, and Bin Chen, Ph.D., and Ira Lubin, Ph.D., Public Health Practice Program Office.
Use of trade names and commercial sources is for identification only and does not imply endorsement by the U.S. Department of
Health and Human Services.References to non-CDC sites on the Internet are
provided as a service to MMWR readers and do not constitute or imply
endorsement of these organizations or their programs by CDC or the U.S.
Department of Health and Human Services. CDC is not responsible for the content
of pages found at these sites. URL addresses listed in MMWR were current as of
the date of publication.
All MMWR HTML versions of articles are electronic conversions from ASCII text
into HTML. This conversion may have resulted in character translation or format errors in the HTML version.
Users should not rely on this HTML document, but are referred to the electronic PDF version and/or
the original MMWR paper copy for the official text, figures, and tables.
An original paper copy of this issue can be obtained from the Superintendent of Documents,
U.S. Government Printing Office (GPO), Washington, DC 20402-9371; telephone: (202) 512-1800.
Contact GPO for current prices.
**Questions or messages regarding errors in formatting should be addressed to
mmwrq@cdc.gov.