 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
| ||||||||||
|
|
|
|
|
Persons using assistive technology might not be able to fully access information in this file. For assistance, please send e-mail to: mmwrq@cdc.gov. Type 508 Accommodation and the title of the report in the subject line of e-mail. Guidelines for Laboratory Test Result Reporting of Human Immunodeficiency Virus Type 1 Ribonucleic Acid DeterminationRecommendations from a CDC Working GroupThe following CDC staff prepared this report: William O. Schalla, M.S. Joann M. Schulte, D.O. in collaboration with the following working group members: Diane P. Francis, M.P.H. Thomas L. Hearn, Ph.D. Terry W. Comans, M.P.A. Harold Dowda, Ph.D. J. Mehsen Joseph, Ph.D. James Versalovic, M.D. Indira K. Hewlett, Ph.D. Summary Monitoring human immunodeficiency virus type 1 (HIV-1) ribonucleic acid levels (also known as HIV viral load) has become the standard of care for monitoring response to therapy in HIV-infected patients. In 1999, CDC published updated surveillance case definitions for HIV infection and acquired immunodeficiency syndrome (AIDS) reporting, including positive results of HIV-1 viral detection tests (CDC. Guidelines for national human immunodeficiency virus case surveillance, including monitoring for human immunodeficiency virus infection and acquired immunodeficiency syndrome. MMWR 1999;48[No. RR-13]:1--28). Since 1996, an increased number of public and private laboratories have begun performing viral load tests. Results obtained with available test methods are variable, and laboratories present these results in different ways, indicating that guidelines to promote standard practice in reporting of test results are warranted. This report provides guidelines for standardized reporting of viral load test results by licensed laboratories to health-care providers and facilities for public health case reporting of HIV infection and AIDS. Recommended standards were developed through data review, input involving a working group of physicians and laboratorians experienced in viral load testing, and an assessment of laboratory practices. These guidelines were discussed, refined, and endorsed at the annual Human Retrovirus and Hepatitis C Laboratory Testing Conference, held March 6--9, 2000, in Charlotte, North Carolina, with participation of representatives from public health, hospital, independent, and blood-collection--facility laboratories. Adoption of these guidelines by all public and private laboratories that perform HIV viral load testing will improve the quality and usefulness of viral load test results for the physician ordering the test and for reporting to public health departments. INTRODUCTIONUsing highly active antiretroviral therapy (HAART) and monitoring therapy response by using viral load testing have contributed to clinical management of persons infected with human immunodeficiency virus (HIV) (1--26). Measurements of viral load and CD4+ lymphocytes are used to determine when antiretroviral therapy should be initiated and to monitor treatment efficacy (27--35). Since 1996, an increasing number of laboratories have been performing viral load tests. In 1999, CDC's Model Performance Evaluation Program (MPEP) conducted a laboratory questionnaire survey, which determined that 51% of the laboratories performing viral load tests had begun doing so in the previous 2 years and 37% had begun in the previous 3--4 years. No test reporting standardization exists; specifically, standard units of measurement of test method have not been established. Laboratory viral load test reports should be accurate and adequate for patient treatment and public health monitoring of the HIV and acquired immunodeficiency syndrome (AIDS) epidemic. To assure test reporting comparability among laboratories, standard methods are needed; moreover, standardized results are needed for early detection of infection, early access to patient care, and early detection of treatment failure. On January 1, 2000, the CDC HIV-infection surveillance case definition was expanded to include viral load test results (34). HIV ribonucleic acid (RNA) viral load reporting is critical in monitoring patients' progression toward AIDS and response to HAART. For public health purposes, viral load reporting is key to expanding surveillance of HIV-infection reporting and improving monitoring of the epidemic. Surveillance programs for HIV/AIDS in the United States, specifically states initiating HIV-infection reporting, will rely on laboratory viral load test reporting to identify prevalent HIV infections among persons who are in care and receiving routine viral load monitoring. Test-reporting standardization will facilitate progress toward electronic laboratory reporting, which is key to increasing efficiency among disease surveillance programs. On November 30, 1999, CDC convened a working group of physicians and laboratorians experienced in HIV testing and representing the Food and Drug Administration (FDA), the Association of Public Health Laboratories (APHL), and the College of American Pathologists to address standardization of laboratory reporting of HIV type 1 (HIV-1) viral load testing results. The working group reviewed information regarding variations in test reporting and interkit variation of test results. At the conclusion of this meeting, the working group developed recommendations, which were discussed, refined, and endorsed at the annual Human Retrovirus and Hepatitis C Laboratory Testing Conference, held March 6--9, 2000, in Charlotte, North Carolina, and sponsored by APHL, with participation of representatives from public health, hospital, independent, and blood-collection--facility laboratories.* This report provides recommendations for standardizing viral load test reports from licensed laboratories to health-care providers and facilities and reporting HIV infection and AIDS cases for public health surveillance. These guidelines do not include a) recommendations for using viral load tests for clinical management, which are reported elsewhere (35,36); b) laboratory safety standards; c) specimen collection, transport, integrity, storage, processing, and analysis; d) data analysis, storage, and transmission; or quality assurance, which is also reported elsewhere (37). BACKGROUNDAvailable Viral Load Tests Three commercially available test kits are routinely used for detecting and quantitating HIV RNA in plasma, including a) the Roche® Amplicor HIV-1 Monitor™ (Roche Diagnostic Systems, Inc.); b) the Bayer Corporation, Diagnostics Division, assay (designated as the Bayer VERSANT® HIV-1 RNA 3.0 Assay [branched chain deoxyribonucleic acid or bDNA], but formerly designated as the Bayer HIV-1 RNA 3.0 Quantitative Assay [bDNA]) and the Bayer Quantiplex™ HIV-1 RNA [bDNA]); and c) the Organon Teknika NucliSens® HIV-1 QT. Of these, the Roche Amplicor HIV-1 Monitor is the only FDA-approved test kit. The methods these kits employ and their range of detection sensitivities are provided in this report (Table 1). The Roche assay is based on a target amplification system and uses reverse transcriptase-polymerase chain reaction (RT-PCR) technology. This assay is available in two versions, 1.0 and 1.5. Version 1.0 is the standard assay and the only FDA-approved assay. The manufacturer has developed a method to increase the sensitivity of the standard assay, and this ultrasensitive method can be used only with version 1.0. Version 1.0 was developed for quantitation of HIV-1 subtype B, the predominant subtype in North America, whereas version 1.5 has been designed to improve equivalent quantitation of non-B subtypes. The Bayer assay, versions 2.0 and 3.0, are based on signal amplification that uses bDNA technology. The Organon Teknika assay is a transcription-based isothermal target amplification method employing nucleic acid sequence-based amplification (NASBA) technology. The NucliSens HIV-1 QT is the present version of the Organon Teknika assay.** Sensitivities of HIV viral load detection vary by test kit type. The Roche standard assay kit has a reportable range of 400--750,000 copies/mL of plasma, whereas the ultrasensitive method has a range of 50--75,000 copies/mL (38). Bayer assay versions 2.0 and 3.0 have a detection range of 500--1,600,000 and 50--500,000 copies/mL, respectively (39). The Organon Teknika's initial NASBA HIV-1 RNA QT detection range was 400--15,000,000 copies/mL, but the NucliSens HIV-1 QT has a detection range of 40--10,000,000 copies/mL (40). Laboratory Practices for HIV Viral Load Testing In 1997, CDC's MPEP implemented an ongoing performance evaluation of laboratories that perform viral load testing. Coded plasma samples of varying RNA levels, which have been obtained from individual*** HIV-1 infected and uninfected donors, are frozen and mailed to approximately 200 MPEP participating laboratories. The laboratories are asked to test the samples in the same manner they test routine or clinical specimens and to provide MPEP with test results, test kit manufacturer information, test control/calibrators/standards results, and quality-control practices. To assess intrashipment reproducibility, selected samples in a panel are duplicated. To assess intershipment reproducibility, all samples in each of two subsequent (i.e., replicate) shipments are identical, except that the vial labeling and sequence are changed so that laboratories will not receive samples containing the same coding or coding within an identical sequence as the original survey. Before each survey, donor samples are characterized by reference laboratories that use kits manufactured by Roche, Bayer, and Organon Teknika. The distribution of laboratory types voluntarily participating in MPEP remained approximately the same during June 1997--February 2000 and included hospitals (52%), independent laboratories (20.5%), health departments (17%), others (10%), and blood banks (0.5%). Approximately 70% of the participating laboratories used the Roche kit in all six survey periods; 22% used a Bayer kit; 6% used an Organon Teknika kit; and 2% used an in-house--developed kit. Of the laboratories using the Roche Amplicor HIV-1 Monitor kit, approximately 25% indicated they had begun using the ultrasensitive procedure. By the last two surveys, all laboratories using the Bayer kit were using the Bayer HIV-1 RNA 3.0 Assay (bDNA); among the nine laboratories using an Organon Teknika kit, all but one were using the NucliSens HIV-1 QT kit. To provide information regarding the variability among laboratory test reporting, results from within test kit manufacturers and within each survey during the six survey periods were examined (Table 2). The median values determined from duplicate sample testing results were reproducible, although variability occurred among the results reported by laboratories using the same test kit and testing the same donor samples (i.e., the duplicate) within a survey or the same donor sample (i.e., the replicate) used in a later survey. When the minimum and maximum values of the reported results were examined for laboratories using the Roche Amplicor HIV-1 Monitor kit, the log difference ranged from 0.8 to 2.2. Similarly, examination of the minimum and maximum reported values for laboratories using the Organon Teknika NucliSens HIV-1 QT kit demonstrated a log difference range of 0.3--0.6. In comparison, a 0.5-log difference occurred within the minimum and maximum reported results from laboratories using the Bayer HIV-1 RNA 3.0 Assay (bDNA), which was the observed difference with all results reported for the same donor sample within and among survey periods (Table 2). Among test kit manufacturers, the median values determined from results reported by laboratories using the Roche Amplicor HIV-1 Monitor and Organon Teknika NucliSens HIV-1 QT were consistently higher than the median values determined from testing results reported by laboratories using the Bayer HIV-1 RNA 3.0 Quantitative Assay (bDNA). This observation was true only for the original sample in a survey and its duplicate. For the same sample used in a later survey (i.e., the replicate), this observation was not true. Instead, good reproducibility**** existed among the values reported for the replicate sample by laboratories using all three manufacturers' test kits. For these surveys, more laboratories use the Roche Amplicor HIV-1 Monitor test kit than the other two kits. If the number of laboratories using the other test kits matched the number of laboratories using the Roche Amplicor HIV-1 Monitor, the range in log difference for results reported by laboratories using the other two test kits would probably increase. Variation Among Laboratory Viral Load Test Reports To understand laboratory practices regarding HIV viral load test reporting, a telephone survey was conducted by San Diego State University (SDSU), under a cooperative agreement with CDC, of randomly selected laboratories from three source groups: medical schools, national commercial laboratories, and laboratories participating in CDC MPEP. Laboratorians who are knowledgeable regarding HIV testing were interviewed, and respondents were asked to fax or mail a copy of a negative and positive HIV RNA report without patient identifiers. A total of 212/279 (76%) telephone surveys were completed; of these, 112 (52.8%) respondents performed HIV RNA testing. Of the respondents performing HIV RNA testing, 3 (2.7%) were blood banks; 69 (61.6%) were hospitals; 11 (9.8%) were health departments; 24 (21.4%) were independent laboratories; and 5 (4.5%) were other laboratory types. Among the 112 respondents, 86 (76.8%) used the Roche Amplicor HIV-1 Monitor; 27 (24.1%) used the Bayer HIV-1 RNA 3.0 Quantitative Assay (bDNA); 11 (9.8%) used the Organon Teknika NucliSens HIV-1 QT kit; and 6 (5.4%) used other methods (e.g., Digene™ and in-house--developed reagents). Multiple test kits were used by 15/112 (13.4%) responding laboratories. A total of 37 different laboratories, 9 (24.3%) independent, 7 (18.9%) health department, 19 (51.4%) hospital, and 2 (5.4%) other laboratories furnished HIV RNA test reports. Of these 37 laboratories, all results were reported in copies/mL; 5 (13.5%) reported in both copies/mL and log10 transformation. Laboratories used different terminology to specify test limits, including the terms linear range, reportable range, sensitivity level, detection levels or limits, and assay limits. Using >1 terms, 26/37 (70.3%) laboratories specified the test kit's lower limits; 12 (32.4%) reported upper limits; and 11 did not provide test limits. One laboratory that specified both a lower limit and an upper limit when results were outside test kit limits did not report the test limits when the results were within the test limit range. When the result was outside the test limits, the report stated that the linear response range for the Roche Amplicor HIV-1 Monitor Test was determined to be within 400--750,000 HIV-1 RNA copies/mL, which is the linear range stated in the manufacturer's insert (38). However, when results were within test limits, the laboratory did not provide the statement indicating the manufacturer's linear range, which indicated information inconsistency among test reports. In certain cases, laboratory slips indicated that HIV had been detected at a value below the test's lower limit (e.g., HIV detected was <400 copies/mL), or the laboratory slip provided an actual number of copies outside of the stated reportable range. The test kit used was reported on the test report form by 9/37 (24.3%) laboratories. Examples of items appearing on the test reports that could introduce difficulty in interpreting test results are provided in this report (Box). RECOMMENDATIONSThe following recommendations were endorsed at the Human Retrovirus and Hepatitis C Laboratory Testing Conference (sponsored by APHL), held March 6--9, 2000, in Charlotte, North Carolina. Good laboratory practice***** requires that a discrete patient identifier be provided to the laboratory with each specimen submitted for retrovirus testing. Because laboratory reporting is critical to public health practice, information regarding date of birth, sex, and racial/ethnic group should be included on the test report form when available. Moreover, the laboratory report should be concise. Required Items To Report The following items must be included on the report form:
Optional Items To Report The following items are optional for test report forms:
Items Not To Be Included on Reports The following items should not be included on the report form:
DISCUSSIONAlthough results from laboratories that use the three commercially produced assays are strongly correlated, the absolute values of HIV viral load measured in the same plasma sample by using two different assays can differ by >2-fold. Data from the MPEP performance surveys demonstrate these differences and illustrate the need for continued surveillance of manufactured tests and laboratory performance. Until a common standard is available to use for normalizing values obtained with different assay methods, choosing one assay method is advisable when HIV RNA levels are monitored to guide therapeutic decision-making. The goal to develop a common standard for normalizing values obtained with different test kits has recently been reported (41). In that study, 26 laboratories, representing 10 different countries, collaborated in establishing the first international standard for HIV-1 RNA that can be used for nucleic acid-based techniques. According to the SDSU survey of laboratory practices, <50% of the laboratories indicated on the results slip what test had been used. Because this information is used to interpret results, whether for clinical care or public health purposes, assay methods and test kit manufacturers should be noted on all viral load test reports. SDSU's survey indicated that all laboratories reported results in copies/mL, and certain laboratories also provided log10 transformation. Although changes can be monitored by using either absolute or log10 values, absolute values are used to determine therapy initiation. In addition, results expressed in both copies/mL and log10 might be useful to health-care providers; therefore, reporting both is strongly recommended. Available tests are not licensed for diagnosing HIV infection, but the viral load test results are used for reporting HIV infection to local and state health departments (34). Although future versions of these tests might be licensed for diagnostic purposes, health-care providers should be aware that available viral load tests are only useful for monitoring clinical status after an HIV diagnosis (35). Available test kits perform differently in detecting non-B subtypes (42--48). In the United States, the Roche Amplicor HIV-1 Monitor version 1.0 is the most popular test for determining viral load. However, it underdetects and underestimates non-B subtypes (e.g., A, E, F, and G) when quantifying viral load (42--47). The Bayer HIV-1 RNA 3.0 Quantitative Assay (bDNA) probably quantifies RNA of different HIV-1 subtypes accurately as a result of redundancy of multiple probes (43--45). The Organon Teknika NucliSens HIV-1 QT assay also underdetects and underestimates divergent subtypes (e.g., A and E) (43--45). All assays have problems amplifying HIV-1 group O and do not amplify HIV-2. The Roche Monitor version 1.5 kit uses a new set of probes and primers that reportedly detect and quantitate non-B subtypes with greater efficiency than the previous version (42--46). Plasma HIV RNA level provides a valid measure of antiretroviral therapy efficacy for HIV-infected persons. Antiretroviral therapy suppression of HIV RNA level followed by rebound levels might signal the emergence of drug-resistant HIV variants, suboptimal adherence to the antiretroviral therapy regimen, decreased absorption of antiretroviral drugs, altered drug metabolism because of physiologic changes, drug interactions, vaccinations, or concurrent infections. An HIV RNA level might be transiently elevated among patients on therapy; the consequences of such elevations is unknown. HIV RNA levels can vary by approximately threefold (0.5 log10) in either direction upon repeated measurements among clinically stable, HIV-infected persons. Changes >0.5 log10 usually cannot be explained by inherent biological or assay variability and likely reflect a biologically and clinically relevant change in the level of plasma HIV RNA. However, plasma HIV RNA assays vary greater toward the lower limits of sensitivity. Thus, differences between repeated measures of >0.5 log10 might occur at low plasma HIV RNA values and might not reflect a substantive biological or clinical change. Viral load reporting has implications for clinical care and HIV/AIDS surveillance. State public health programs are implementing recommendations by the Council of State and Territorial Epidemiologists and CDC for nationwide HIV surveillance, including reporting detectable RNA viral loads (34). As part of this reporting, laboratories and health-care providers would be required to report HIV-infection cases to state and territorial health departments. Standard methods are needed for viral load test reporting to providers and health departments so that clinicians and public health professionals will have a common language to use in conducting patient care and monitoring the epidemic. To facilitate developing that common language, CDC and the Council of State and Territorial Epidemiologists recommend that all states adopt rules or regulations to require reporting of detectable viral loads to public health departments. One state requires reporting of undetectable viral load results based on the lower limit sensitivities of the manufactured kits, but then follows up with health-care providers to document whether the patient has other test results or clinical evidence indicative of infection with HIV. Reporting of viral load test results by using standard nomenclature, test results presentation, and electronic data transfer will permit reporting of HIV cases in a way that is compatible with electronic laboratory efforts being developed for national notifiable disease reporting (49). References
*The conference report can be ordered from APHL at Internet: <http://www.aphl.org> (accessed July 19, 2001) **Certain enzyme immunoassay (EIA or enzyme-linked immunosorbent assay [ELISA]) screening and Western blot confirmatory tests used in the United States reliably detect HIV-1 infection but not HIV-2 infection. HIV infections acquired in Africa, specifically West Africa, might be HIV-2 infection and escape detection if assays optimized for HIV-1 are used. Health-care providers who perform HIV tests on patients who might have been exposed in Africa (e.g., returning visitors or newly arriving immigrants and refugees) should use EIAs that are licensed for detection of both HIV-1 and HIV-2. Additionally, the majority of commercially available tests that are used to detect HIV RNA viral load are effective for HIV-1 subtype B but not other HIV-1 subtypes (e.g., A, C, D, or E). No FDA-approved tests for quantification of non-B HIV subtypes exist. HIV infections acquired outside the United States might be HIV-1 non-B subtypes. Therefore, when testing patients for viral load who might have been exposed in Africa or other overseas locations, assays that are effective for multiple HIV-1 subtypes (HIV-2 RNA viral load tests are not commercially available) should be used. ***Samples are obtained from individual donors; samples from different donors are not pooled, and individual samples are not diluted. ****Good reproducibility can be defined as acceptable testing performance of duplicate and replicate samples influencing the quality of precision and accuracy (Sources: Taylor JK. Principles of quality assurance of chemical measurements. Gaithersburg, MD: US Department of Commerce, National Bureau of Standards, 1985; publication no. NBSIR 85-3105; and Taylor JK. Quality assurance of chemical measurements. Anal Chem 1981;53:1588A). *****Good laboratory practice is defined as an acceptable way to perform a basic activity that is known to influence the quality of its output (Source: Taylor JK. Good laboratory and good measurement practices. In: Quality assurance of chemical measurements. Chelsea, MI: Lewis Publishers, Inc., 1987;112--3.) Table 1  Return to top. Table 2 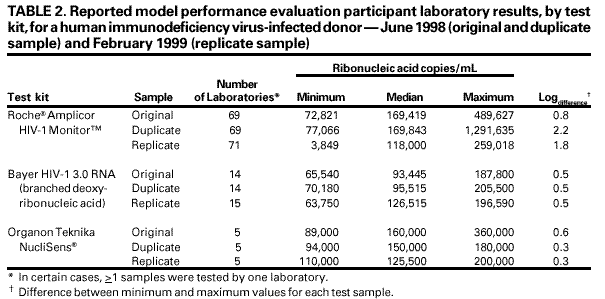 Return to top.
All MMWR HTML versions of articles are electronic conversions from ASCII text into HTML. This conversion may have resulted in character translation or format errors in the HTML version. Users should not rely on this HTML document, but are referred to the electronic PDF version and/or the original MMWR paper copy for the official text, figures, and tables. An original paper copy of this issue can be obtained from the Superintendent of Documents, U.S. Government Printing Office (GPO), Washington, DC 20402-9371; telephone: (202) 512-1800. Contact GPO for current prices. **Questions or messages regarding errors in formatting should be addressed to mmwrq@cdc.gov.Page converted: 11/16/2001 |
|||||||||
This page last reviewed 11/16/2001
|
